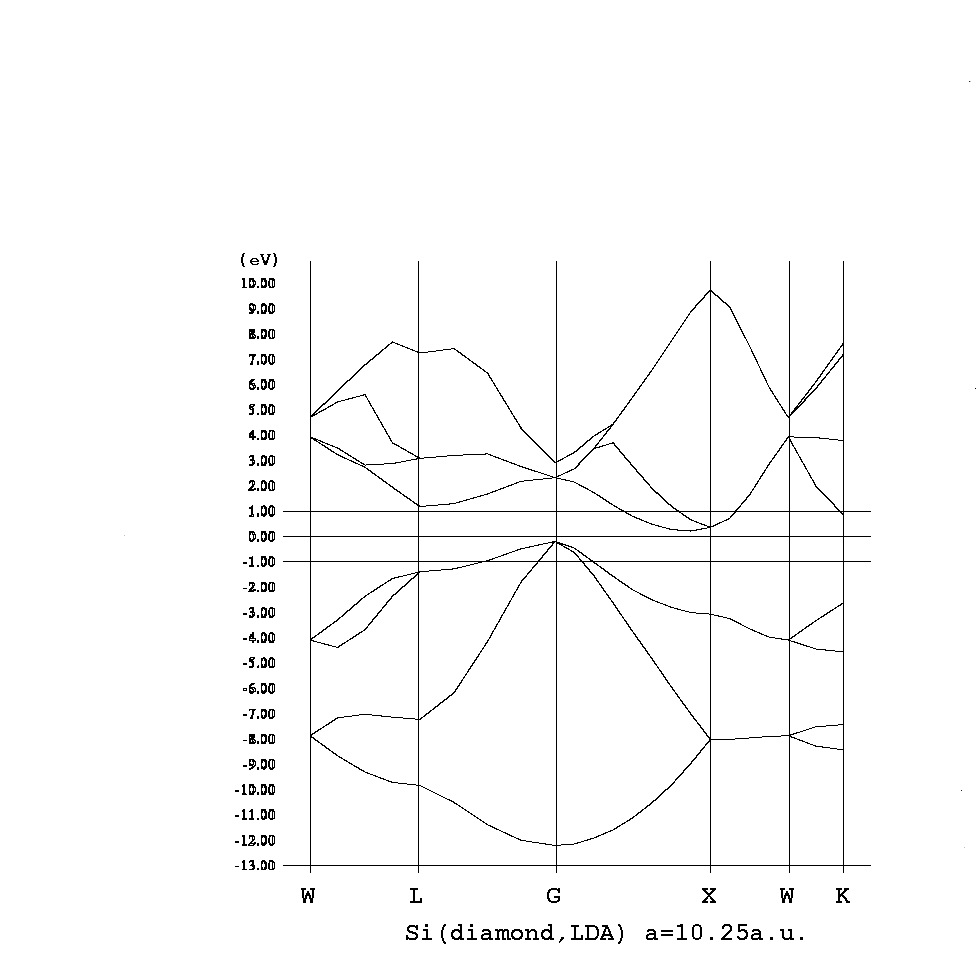
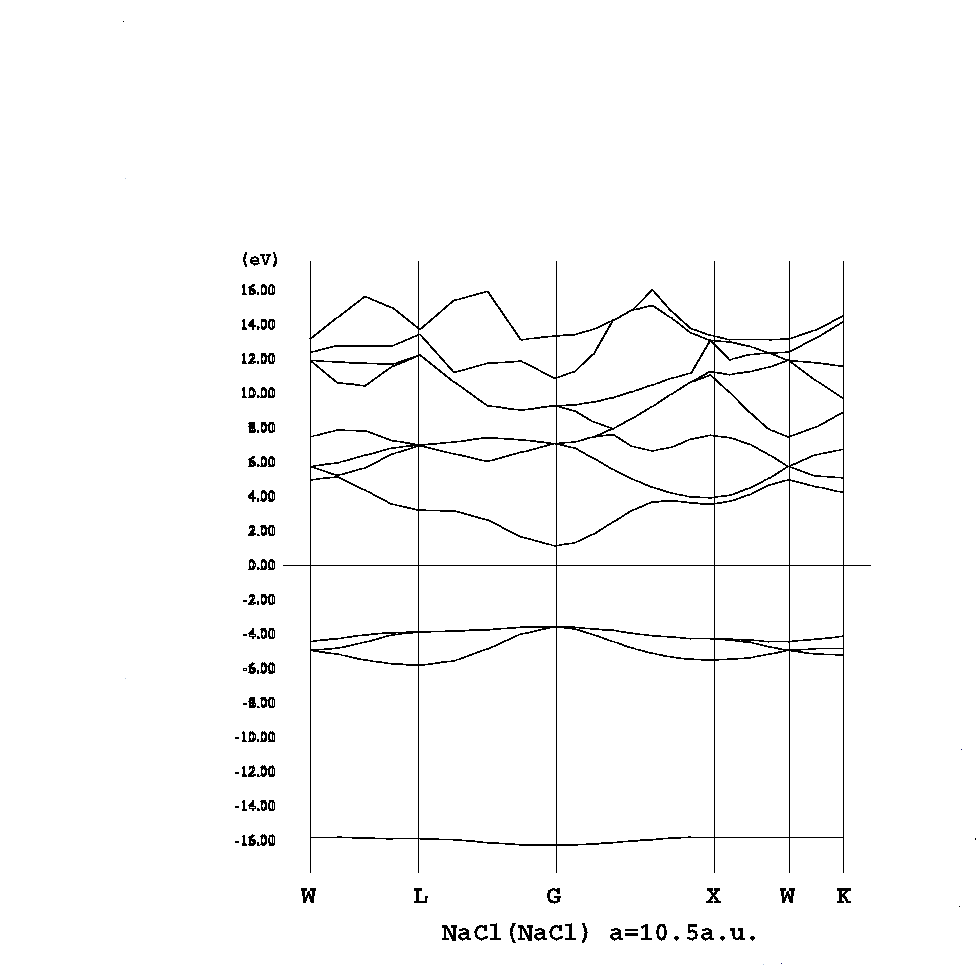
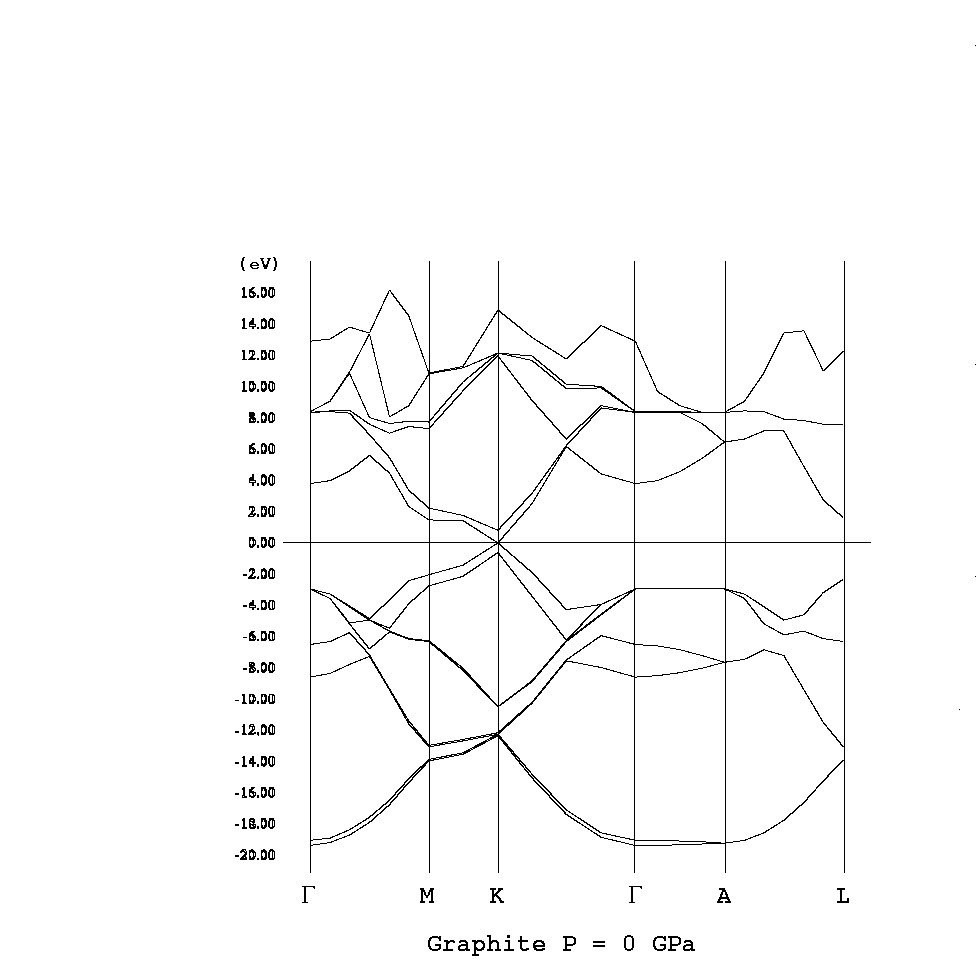
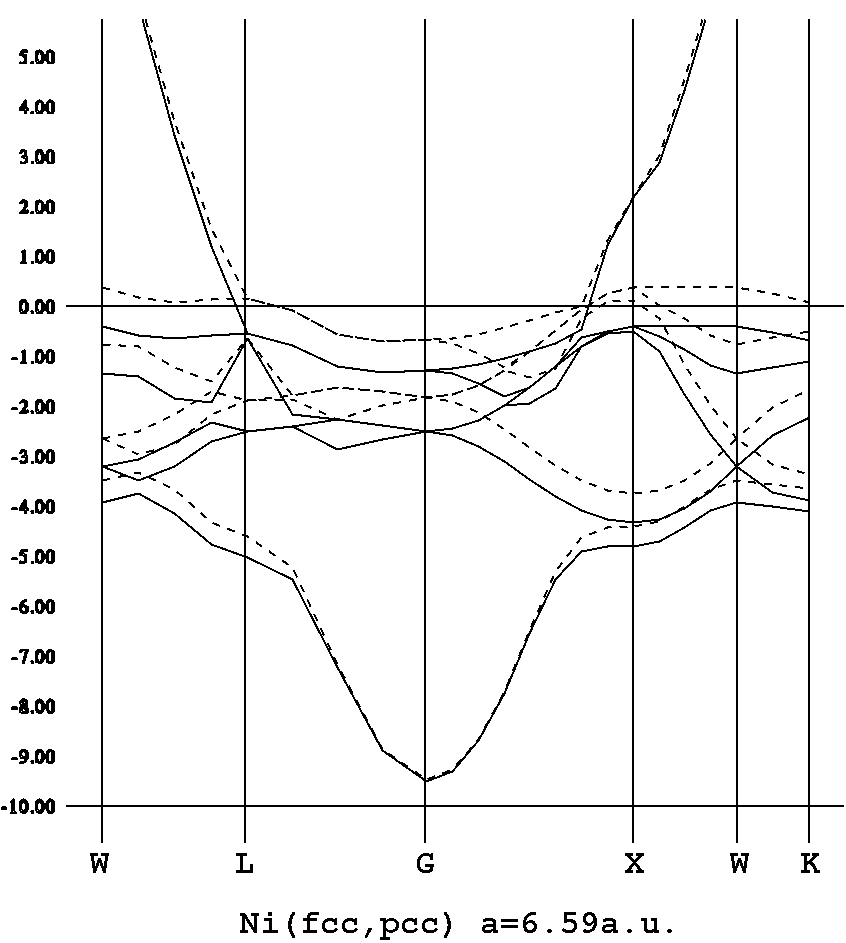
コーン・シャム方程式
(コーン・シャムの式、KS方程式、Kohn-Sham equation)
Kohn-Shamの論文(W. Kohn and L. J. Sham, Phys. Rev. 140,
A1133(1965))で初めて登場した、一電子近似下での電子問題を解くためのシュ
レーディンガー方程式様の式。
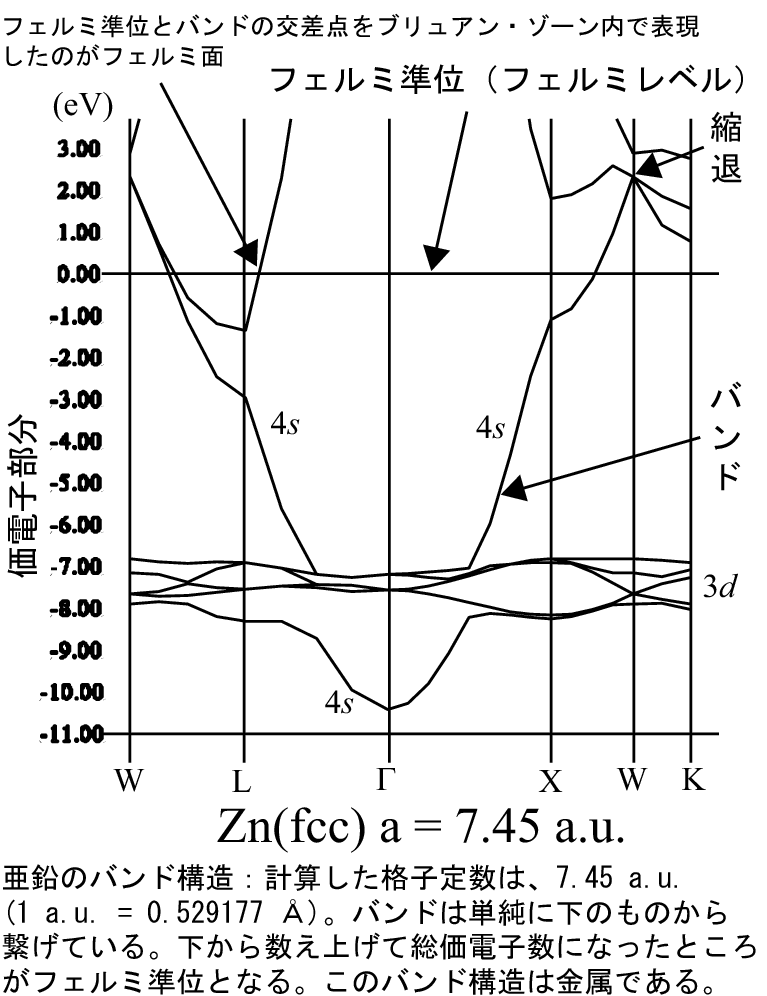
HΨi = εiΨi, H = -1/2Δ + Vext + VH + Vex
Vext + VH + Vex ← 一体(一電子)有効ポテンシャル部分
ρ(r) = Σi|Ψi(r)|2
ρ(r):電荷密度、Ψi(r):波動関数(iはバンド指標、本当はk点に関しての指標
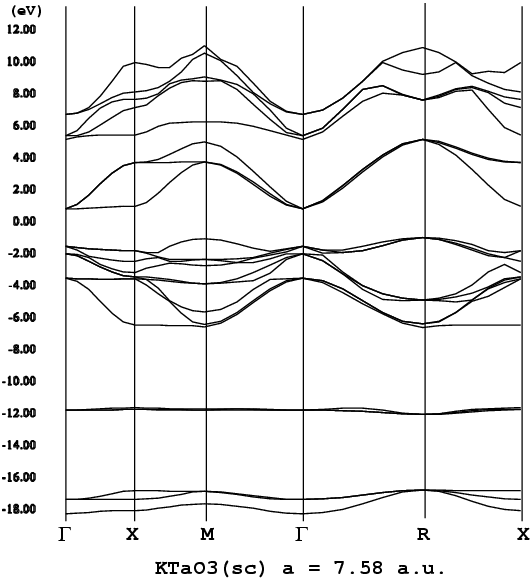
もあるが省略)、εi:軌道エネルギー(ε
i,kとするとバンド構造を描け
る、k:BZ内のサンプリング点、つまりk点)、Vext、
VH、Vex:それぞれ、外場ポテンシャル、ハートリー
(電子間クーロン)ポテンシャル、交換・相関ポテンシャル。スピンはここで
は考えない。またエバルト項なども考えない。
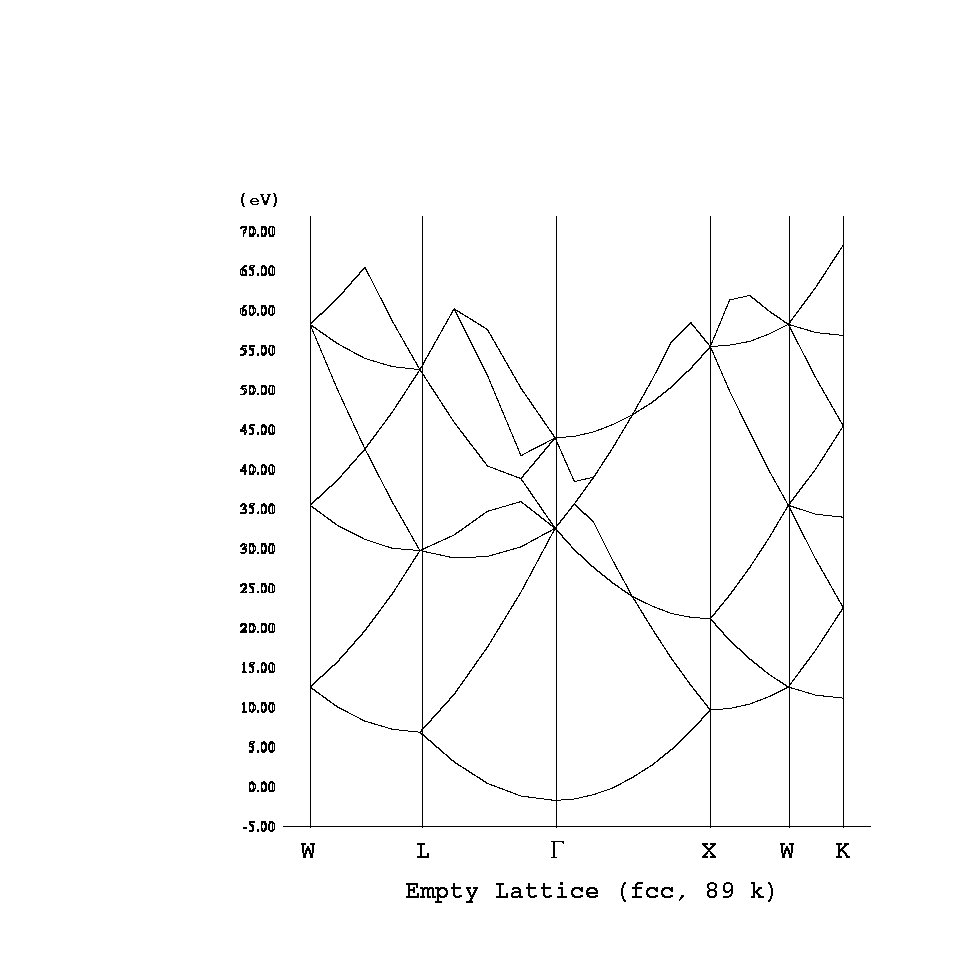
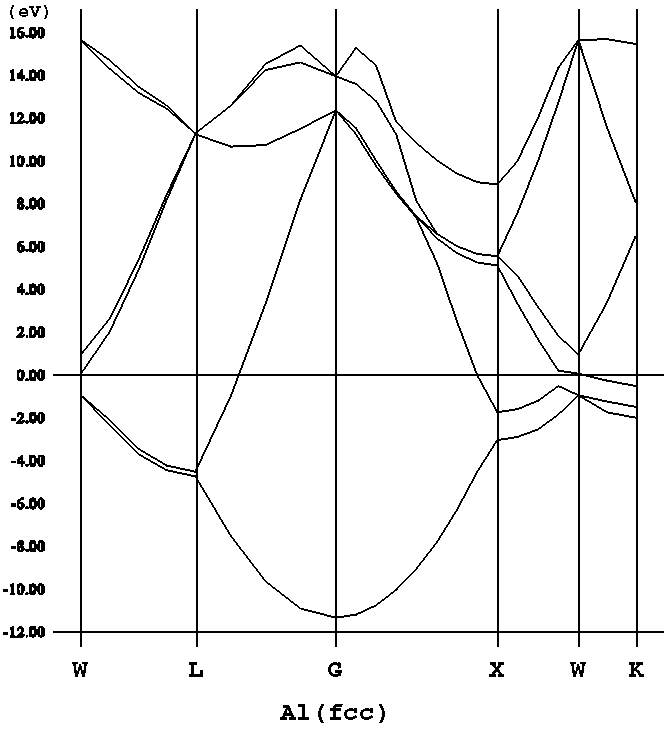
KS方程式のポテンシャル部分を取ってしまうと、H = -1/2Δだけとなり、自
由電子模型に対するシュレーディンガー方程式となる。←運動エネルギー部分
が、自由電子(=相互作用していない)のものになっていることも重要。→
交換・相関部分(Vex)に押し込める。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}