とても最近に、やらかした計算上のバグ、失敗
[Top][失敗知識データベース
(畑村創造工学研究所←JSTから移動)]
- (れ)本日のバグ(レポート6/2、2026)
- (た)本日のバグ(レポート9/13、2024)
- (よ)本日のバグ(レポート3/28、2024)
- (か)本日のバグ(レポート4/6、2022)
- (わ)本日のバグ(レポート10/15、2020)
- (を)本日のバグ(レポート8/28、2019)
- (る)本日のバグ(レポート1/8、2019)
- (ぬ)本日のバグ(レポート11/20、2017)
- (り)本日のバグ(レポート12/4、2015)
- (ち)本日のバグ(レポート5/22、2015)
- (と)本日のバグ(レポート12/3、2014)
- (へ)本日のバグ(レポート12/4、2013)
- (ほ)本日のバグ(レポート8/23、2013)
- (に)本日のバグ(レポート5/31、2013)
- (は)本日のバグ(レポート3/22、2013。5/16、2013追記)
- (ろ)本日のバグ(レポート6/26、2012)
- (い)本日のバグ(レポート10/13、2011)
- (Z)本日のバグ(レポート1/21、2011)
- (Y)本日のバグ(レポート3/2、2010)
- (X)本日のバグ(レポート9/4、2009。12/4、2009追記)
- (W)本日のバグ(レポート8/5、2008)
- (V)本日のバグ(レポート2/8、2008)
- (U)本日のバグ(レポート12/17、2006)
- (T)本日のバグ(レポート8/21、2006)
- (S)本日のバグ(レポート9/16、2005)
- (R)本日のバグ(レポート2/11、2005)
- (Q)本日のバグ(レポート9/26、2005)
- (P)本日のバグ(レポート1/12、2005)
- (O)本日のバグ(レポート2/25、2004)
- (N)本日のバグ(レポート4/9、2003)
- (M)本日のバグ(レポート12/5、2002)
- (L)本日のバグ(レポート12/19、2001)
- (K)本日のバグ(レポート7/16、2001)
- (J)本日のバグ(レポート3/2、2001)
- (I)本日のバグ(レポート9/18、2000)
- (H)本日のバグ(レポート8/29、2000)
- (G)本日のバグ(レポート5/25、2000)
- (F)本日のバグ(レポート2/24、2000)
- (E)本日のバグ(レポート1/17、2000)
- (D)本日のバグ(レポート4/20、1999)
- (C)本日のバグ(レポート1/20、1999)
- (B)本日のバグ(レポート11/18、1998)
- (A)本日のバグ(レポート11/2、1998)
- (9)本日のバグ(レポート8/4、1998)
- (8)本日のバグ(レポート3/17、1998)
- (7)本日のバグ(レポート1/20、1998)
- (6)本日のバグ(レポート12/5、1997)
- (5)本日のバグ(レポート10/15、1997)
- (4)本日のバグ(レポート6/18、1997)
- (3)本日のバグ(レポート3/26、1997)
- (2)本日のバグ(レポート2/6、1997)
- (1)本日のバグ(レポート1/23、1997)
(れ)本日のバグ(レポート6/2、2026)[目次]
よくある間違いをまたやらかす。コメントアウトした文(注釈行)を、元に戻す(注釈を外す)時、その文はDOループの外にあった。但し、当該文は、本来DOループ内にあるべきものだった。それに気付かず、そのまま実行しておかしな結果を返した。結果が明かに妥当でなかったおかげで、間違いにすぐに気付くことができた。微妙なちがいうだったら、間違いを認識できなかった可能性があった。今後も気を付ける必要がある。
(た)本日のバグ(レポート9/13、2024)[目次]
今回の間違いは、バグと言うより単純な設定の誤りである。構造最適化における、ちょっと特殊な場合に対する設定を、通常の場合のものに戻すことを失念し、その特殊な設定のまま構造最適化をしたため、おかしな結果となっていた。割とよくありそうな単純ミスである。
(よ)本日のバグ(レポート3/28、2024)[目次]
力の大小の判定において、力がベクトルであることを考えずに判定するプログラムを考え、間違った計算を行っていたことが判明した。ここでのベクトルの大小判定は、そのノルム(大きさ)だけを考えればよかったので、絶対値として考えればよかった。
つまり、
F > 0.001
ではなく、
|F| > 0.001
とするべきだった。当初、絶対値としなかったため、力が負の値の場合(力はベクトルなので負の値が存在)が考慮されず、計算が正しく進まなかった。
修正後、計算結果の推移は妥当並ものとなった(5/17、2024)。
(か)本日のバグ(レポート4/6、2022)[目次]
ループ内に書くべき構文を、ループの外に記述したため、間違った計算をしてしまう。
DO 20 K=1,NK
構文1
構文2
.
.
.
構文X(C = C + D(K))
20 CONTINUE
とすべきところを、
DO 20 K=1,NK
構文1
構文2
.
.
.
20 CONTINUE
構文X(C = C + D(K))
としてしまった。このため構文Xにあった、ループ内の変数を使った変数の部分(D(K))が未定義となり、おかしな結果を返すこととなった。構文Xをループ内に記述することで、正しい結果を返すようになった。
構文Xの部分は後から追加したもので、追加する時、(本当はループ内で追記すべきところを)ループ外に記述して、このような事態になった次第である。
(わ)本日のバグ(レポート10/15、2020)[目次]
座標変換の仕方を間違えて、誤った構造のまま計算を続けてしまう。
事の発端は、バルクでの計算から表面計算に移行するために、単純に単位胞の高さ方向を2倍にして、セル(胞)1個分を真空層としたスラブモデルを考えた。この場合、座標は規格化された数値を用いれば、0.5 → 0.25となる。0.25なら、0.125となる。ここで筆者が間違えたのは、-0.25を、-0.125としたことである。周期的境界条件から、-0.25 = 0.75である。便宜上、-0.25として扱う場合があった。因みに、-0.25としても、0.75としてもどちらも同じ結果を与えることは確認・検証済である。で、-0.25 → -0.125は間違いで、正しくは、-0.25 → 0.75 → 0.375 (或は、-0.625 = -0.125 + (-0.5))である。
筆者は、大分計算を進めた後で、構造最適化したスラブ表面を描画して、構造が非対称でおかしいことに気付いた。更に、まだ問題点があった。それは、原点上の原子の扱いである。周期的境界条件では、座標が0.0は、1.0と等価である。従って、スラブモデルでないバルクの場合、0.0のみを使用し、1.0の方は扱わない(そうしないと二重に計算されてしまう)。これに対しスラブで扱う場合、最早バルクで1.0だった座標は、セル(胞)の中の原子となるため、1.0 → 0.5として扱う必要がある(原点とは別の原子として扱う)。つまりこの場合、スラブとして扱うために0.5に相当する原子1層分を追加して考える必要がある。実は、これもスラブ仔構造を描画したものが対称になってないことで気付いた。
(10/20、2020)修正後の計算結果は、力の値が構造(とその対称性)を反映した正しいものとなった。
(を)本日のバグ(レポート8/28、2019)[目次]
今回の失敗は、バグではない。対称性から明かにおかしな力の値が出ていて、どうしてかと試行錯誤した結果、単に計算が収束していなかったのが原因だった。より簡単なテストケースでは、系の持つ対称性に反しない正しい力が得られたのに、本番では全くおかしな力となっていた。筆者は、力の値にのみ着目したため、それ以外の問題点に暫く気付くことができなかった。ふと、計算結果同士の比較で全エネルギーを見ていたら、テストケースでは十分収束していた全エネルギーが、(力がおかしなケースでは)全く収束していないことに気付いた。
で、計算が収束するようにパラメータ(この場合は、電荷の混合の比)を調整すると、無事計算は十分に収束し、力も系の持つ対称性を反映したものとなった。今回も思い込みで、本当ならもっと早く気付くことが可能であった問題点を見過ごしていた(猛省)。
(る)本日のバグ(レポート1/8、2019)[目次]
新しい系の計算をしようとして、過去に計算した他の系の計算プログラム、データを流用したが、得られた結果がおかしかった。構造最適化(対称性の高い系だったので格子定数のみが対象)する場合、普通全エネルギーは下って(低くなる、小くなる)いくのだが、今回の場合、上っていってしまった。つまり構造最適化するほどエネルギー的に不安定になっていった。
正しい結果を与えることが確実なプログラムで検証してみると、予備的な結果ながら全エネルギーには問題ないことが判明した。更に検証するとストレスの計算結果がおかしいことが判明した。そこで不具合を示すプログラムを調べると、ストレス計算部分で特殊な条件を課して計算していることが判明した。この計算は、対称性を課していない場合には妥当な結果となるように施してあり、対称性を課している場合には全く間違った結果を与えるものだった。筆者は、そのことを忘れて、対称性を課した系でこのプログラムを使用し、おかしな結果を得ていた。
早速、当該条件を課すことをやめて、計算し直した結果、正しい結果を与えるようになった。
(ぬ)本日のバグ(レポート11/20、2017)[目次]
プログラムを拡張して、計算式を記述したが、記述した式に本来あるべき、DCONJGがなくて、正しい結果を与えなかった。元の計算式を記述した部分には、DCONJGが正しく記述されていたにもかかわらず、これに気付かなかった。バンド計算では、多くの場合、A*・Aのように複素共役したものとのかけ算(例:電荷密度は波動関数のノルム:ρ(r) = Ψ*(r)・Ψ(r))が行われており(今回もそうだった)、それに気付かずそのまま計算していた。
原因を突きとられたのは、全エネルギーの値が異っていたが、フェルミレベルの数値が一致していたこと。そこで全エネルギー関連の記述をあれこれ調べている内に、問題点に気付いた次第。
それは、筆者の[D論](PDF形式、320 kb)にある全エネルギーの(2.42)式の、右辺第2項以降の部分(複素共役がたくさんある)が関係している。SCF計算において、これらの部分は影響しないので、全エネルギーの値は違っても、フェルミレベルの値は一致する場合がある(←そこから問題点を絞り込める)。今回、単純にこれら全エネルギーの項と同様な新しい計算部分を付加した時、DCONJGを付け忘れたことが原因だった。
今回は、上記以外にも、double count(二重で足し込む)していた部分があった(半分にするための正しい係数を付けていなかった)。これは、力の計算に影響した。修正後、正しい結果を返すことを確認した。この問題の発見は少し時間がかかった。部分内殻補正(PCC)に関る部分での問題だったので、同補正が必要ない計算では、問題が顕在化しなかったからである。
(1/10、2018)更に、サブルーチンの引数の不整合によるエラーも発見した。これまで、引数の不整合が顕在化しない条件で計算していたが、新しい条件で計算すると、途端に”セグメンターションがおかしい”と言て、実行が止った。そこで調べてみると、サブルーチンをCALLする時と、サブルーチン自身の引数とが整合していなかった。
(り)本日のバグ(レポート12/4、2015)
[目次]
今回は、k点のメッシュについてです。メッシュの取り方によって一見正し
そうに見えて実は問題がある例です。
メッシュの刻みが少ない場合、扱うk点が全て対称性の良い点だけとなり、
対称性の低い一般点がない場合があります。そのような場合、よりメッシュ刻
み数が多い場合(対称性の低い一般点などが入ってくる場合)と挙動が異なる
ことがあります。以下、k点メッシュの参考例(図)。
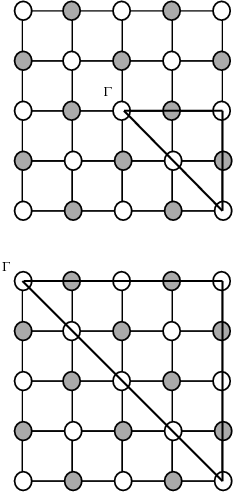 (png画像、7.5 kb)
(png画像、7.5 kb)
上側が少ないメッシュ(3×3相当)、下側がより細かいメッシュ(5×5
相当)。本当は、3×3×3、5×5×5と3次元を想定しています。太い幅
の線によって示される三角形の部分が、計算対象となるブリュアンゾーン(BZ)です。少ないメッシュ
では、線上にしかk点(○で表現、○と●は特に違いがある訳ではない)が存
在しません。つまり対称性の高いk点しかない。一方、下側には、対称性が高
くない一般のk点も存在します。
3×3(×3)の場合、三角形ではなく、それが二つ合わさった四角形(正
方形)として(バンド計算して)も、対称性から結果に差が出ない場合があり
ます。一方、3×3(×3)で同じ結果でも、メッシュを5×5(×5)にす
ると、三角形と正方形では同じ結果にならない場合があります。
本当に正しいかどうかの検証は、P1対称性(恒等変換のみ)の場合、つまり
BZ全体でメッシュをとった結果と比較します。従って、上記の三角形、正方形
の場合、対称性を考慮して計算しています。P1対称性の結果と一致すれば、そ
れは正しいと判断できます。ただどの場合も、メッシュが同じになっている必
要があります。三角形の時は、3×3で、BZ全体では、それとは異なる刻みで
メッシュを取った場合、結果は異なってしまいます。
(ち)本日のバグ(レポート5/22、2015)
[目次]
本日のバグは、プログラム改良(と言うか拡張)時に発生しました。それは、
B = DCONJG(A(10))*A(10)
を、本来、
B = DCONJG(A(IA))*A(IA)
とすべきとろこを、
B = DCONJG(A(10))*A(IA)
としてしまいました。DCONJG(A(IA))*A(IA)と正しく修正していれば、Bの値
は正の数値になるのですが、実際に出てきた値は、正負両方でした。修正した
はずのプログラムを良く見たら、DCONJG(A(10))*A(IA)とあり、変更が中途半
端になっていた訳です。DCONJG(A(IA))*A(IA)にして問題は解決しました。
(追記:5/29、2015)
今回の失敗は、上記とは別のものですが、さして大きな失敗(と言っても侮
ることはできない)ではないので、”追記”という形にしました。
で、それは計算結果の足し上げ(積分)において、足し上げの範囲(積分範
囲)を間違えて計算していました。その計算結果は状態密度で、フェルミレベルまで足せば、
その状態の総数(扱ったユニットセル中の全電子数)になります。ところが、
実際の計算では、フェルミレベルではなく、扱ったエネルギーの全範囲(つま
りフェルミレベルより上の状態も足し上げる)を足し上げて(積分して)いま
した。そして、それによって得られた値を系の全電子数(ユニットセル内の全
電子数)として規格化等の計算を行なっていました。これは明らかにおかしな
計算になっています。
この間違いに気付いた後、直ちに修正を行ない、計算をやり直しました(5/
29、2015段階では、やり直し計算の最中)。
(と)本日のバグ(レポート12/3、2014)
[目次]
本日のバグ(失敗)は、WRITE文による出力での問題です。状況としては本
当に単純なものですが、場合によっては深刻な影響を与える可能性があります。
実は、この手の失敗は、過去にも何度となく犯していました。今回の失敗の状
況は、概して以下のようなものです。
SCF-Loop
DO Loop
WRITE文
END DO Loop
END SCF Loop
筆者としては、SCF計算(SCF:Self-Consistent Field、自己無撞着場)が収
束した段階での値を出力を望んでいました。しかし漫然として作成したプログ
ラム上では、上記のようにWRITE文による出力を”漫然”とさせてしまいまし
た。このため、イタレーションの第一回目から出力され続けたため、出力先の
ファイルの容量が膨大なものになってしまいました(筆者の計算手法では、
SCF計算の収束に割と多くの繰り返し計算が必要だったため尚更だった)。容
量がHDD(ハードディスク)の書き込み制限量をオーバーすると計算が止まっ
てしまいます。
以下のように、「収束(Converge)したらWRITE文で出力する」とすれば、問
題はなくなります。
SCF-Loop
DO Loop
IF SCF calculation Converged THEN
WRITE文
END IF
END DO Loop
END SCF Loop
或いは、収束した後(SCF Loop終了後)、別途WRITE文で出力させても良い
です。以下のやり方では、SCF Loop外へ、WRITE出力するための配列等を保持
しておく必要があります(場合によってメモリがその分損になります)。やり
方としては他にもいろいろあるはずです。
SCF-Loop
DO Loop
いろいろ計算
END DO Loop
収束判定
END SCF Loop
DO Loop
WRITE文
END DO Loop
今は、HDDの容量も増し、ユーザー一人当たりの使用可能ディスク領域の容
量も飛躍的に大きくなっています(100 GBくらいは標準的?)。昔なら、この
手の失敗で、個人単位での書き込み可能なディスク領域を溢れさせて、場合に
よってはシステム全体に影響を与えることも多々あったりしました(筆者も時
たまやらかした)。今は、容量的にそうそうないとは言えますが、気を付ける
に越したことはありません。
(へ)本日のバグ(レポート12/4、2013)
[目次]
バグ(や失敗)はなくならない。今回は、良く見たらコードにミス(誤り)
があった例です。以下、当該コード部分(抜粋)です。
OPEN(UNIT=91,FILE='./BAND/Ni001/C.61')
OPEN(UNIT=92,FILE='./BAND/Ni001/C.62')
OPEN(UNIT=93,FILE='./BAND/Ni001/C.63')
OPEN(UNIT=94,FILE='./BAND/Ni001/C.64')
OPEN(UNIT=95,FILE='./BAND/Ni001/C.65')
OPEN(UNIT=96,FILE='./BAND/Ni001/C.66')
OPEN(UNIT=97,FILE='./BAND/Ni001/C.67')
OPEN(UNIT=99,FILE='./BAND/Ni001/C.68')
OPEN(UNIT=100,FILE='./BAND/Ni001/C.69')
OPEN(UNIT=101,FILE='./BAND/Ni001/C.70')
OPEN(UNIT=102,FILE='./BAND/Ni001/C.71')
上記は、ファイルに関するOPEN文の記述ですが、深
紅色の部分そのものは間違いではありません。問題は、当該行
("UNIT=97")の次のUNITが、"UNIT=99"になっていることです。本当は、そこは
"UNIT=98"になるべきものでした。この欠落によって、"UNIT=98"相当する部分
の後の計算と、ファイル操作も行なわれません。システムによりますが、筆者
の使用するマシンでは既定値としてfort.98がカレントのディレクトリ上に作
られます。上記のコードにあるように本来なら、指定したディレクトリ上に、
"C.68"("UNIT=99"以降は、C.68 → C.69、C.69 → C.70...と変更される)が
作られるはずのものでした。
筆者は、この間違いに何年も気付きませんでした。偶然当該コードを扱うこ
ととなり、コードを修正している途中で、数字の対応(上記コード上の数字の
一の位の部分)に違和感を感じて、おかしいことに気付きました。
(影響)調べた結果、この誤りによる影響は、ほとんどの場合で問題ないこ
とが分かりました。多くの場合、実際には使用や利用しない部分でした。逆に
そのため、間違いに気付くことが出来なかったと言えます。
(ほ)本日のバグ(レポート8/23、2013)
[目次]
今回のバグは、力に関するものです。症状としては、表示される力の値が、
計算途上で発散します。ただ、この症状が出る計算では原子は動かしません
(力は使わない)。この”動かさない”ことがバグの原因になっています。た
だ、原子を動かさない(単位胞内部の原子の構造最適化を行なわない)ため、
この力の発散は計算結果に影響を与えません。
何故、このような事態になったかというと、単純に初期化をしなかったから
です。筆者の計算コードでは、力の計算を行なうサブルーチンが主に2つ(FORCE
とFORLOC)
あり、サブルーチンFORCEでのみ力の初期化をしていました。で、力の計算の
必要がない時、このサブルーチンFORCEは呼び出されず、FORLOCのみ呼び出さ
れていました(力の計算をしない場合、本当はFORLOCも呼び出す必要はありま
せん)。従って、SCF計算(SCF: Self-Consistent Field)が繰り返され、
FORLOCが呼び出される毎に、初期化されない力の値がどんどん足されていき
(当然、本当は力を再計算する場合、ゼロ初期化しておく必要があります)、
値が発散してしまった訳です。
つまり、力の計算をする場合、
-SCF-LOOP START
|
| CALL FORCE
|
| CALL FORLOC
|
| WRITE (6,*) FORCE-VALUE
|
-SCF-LOOP END
で何ら問題ありません。一方、力の計算を行なわない場合でも、
-SCF-LOOP START
|
| NOT CALL FORCE
|
| CALL FORLOC
|
| WRITE (6,*) FORCE-VALUE
|
-SCF-LOOP END
サブルーチンFORCEは呼ばないのに、サブルーチンFORLOCは呼んでいて、か
つ力の値も表示していました。FORLOC内では、FORCE-VALUE = FORCE-VALUE +
ADD-FORCEと足し込みを初期化しないで行なっていました。このため、
SCF-LOOP中、FORLOCをCALL(呼び出す)毎にこの足し上げがどんどん加算され
ていき、発散した値が表示された訳です。
そもそも、力の計算が必要ないのに、FORLOCを呼び出して、加えて漫然と力
の値を表示させること(力の計算が必要ない時でも、値を表示させる必要があ
る事例は存在します)に問題がありました。
以下、力の発散の様子。原子は理想位置なので力はゼロとなります。実際、
最初の計算では正しくゼロです。この最初の計算でのみ初期化が行なわれてい
ます。最初は、小さな値でしたが、あっというまに大きな値になり、発散
("*"は値が大き過ぎて表示不能の意)しています。
ITER = 1001
1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
2 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
3 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
4 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
5 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
6 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
7 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
8 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
9 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
10 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
11 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
12 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
13 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
14 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
15 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
16 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
ITER = 1041
1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
2 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
3 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
4 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
5 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
6 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
7 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
8 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
9 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
10 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
11 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
12 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
13 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
14 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
15 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
16 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
ITER = 1081
1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
2 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
3 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
4 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
5 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
6 0.0000001 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
7 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
8 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
9 0.0000001 0.0000000 -0.0000002 0.0000000 -0.0000002 0.0000000
10 0.0000000 0.0000000 0.0000002 0.0000000 0.0000003 0.0000000
11 -0.0000001 0.0000000 0.0000001 0.0000000 0.0000003 0.0000000
12 -0.0000001 0.0000000 -0.0000001 0.0000000 -0.0000004 0.0000000
13 -0.0000001 0.0000000 -0.0000001 0.0000000 0.0000000 0.0000000
14 0.0000003 0.0000000 -0.0000001 0.0000000 0.0000001 0.0000000
15 0.0000003 0.0000000 0.0000000 0.0000000 -0.0000001 0.0000000
16 0.0000001 0.0000000 -0.0000001 0.0000000 -0.0000001 0.0000000
ITER = 1121
1 0.0000000 -0.0000143 0.0000000 0.0000239 0.0000000 0.0000097
2 0.0000000 -0.0000333 0.0000000 0.0000030 0.0000000 -0.0000119
3 0.0000000 -0.0000129 0.0000000 -0.0000058 0.0000000 0.0000129
4 0.0000000 -0.0000025 0.0000000 0.0000124 0.0000000 0.0000070
5 0.0000000 -0.0000420 0.0000000 -0.0000225 0.0000000 0.0000268
6 0.0000000 0.0000543 0.0000000 0.0000152 0.0000000 -0.0000094
7 0.0000000 0.0000316 0.0000000 0.0000028 0.0000000 -0.0000069
8 0.0000000 -0.0000007 0.0000000 -0.0000133 0.0000000 0.0000059
9 0.0000000 0.0000888 0.0000000 -0.0001653 0.0000000 -0.0001897
10 -0.0000001 -0.0000462 0.0000000 0.0002063 0.0000000 0.0002982
11 0.0000000 -0.0000765 0.0000000 0.0001060 0.0000000 0.0002732
12 -0.0000001 -0.0000818 0.0000000 -0.0000730 0.0000000 -0.0004109
13 -0.0000001 -0.0001103 0.0000000 -0.0000580 0.0000000 0.0000076
14 -0.0000001 0.0003215 0.0000000 -0.0000697 0.0000000 0.0001188
15 -0.0000001 0.0003413 0.0000000 -0.0000345 0.0000000 -0.0000558
16 -0.0000001 0.0000681 0.0000000 -0.0001443 0.0000000 -0.0000923
ITER = 1161
1 0.0148666 -0.0000168 -0.0247797 0.0000001 -0.0100842 0.0000005
2 0.0345371 -0.0000264 -0.0031422 -0.0000015 0.0123460 -0.0000012
3 0.0133288 -0.0000083 0.0060150 0.0000014 -0.0133701 0.0000011
4 0.0025655 -0.0000220 -0.0128929 -0.0000015 -0.0072616 -0.0000012
5 0.0435534 -0.0000197 0.0232800 0.0000014 -0.0277751 0.0000010
6 -0.0563418 0.0000062 -0.0157138 -0.0000004 0.0097173 -0.0000004
7 -0.0327284 -0.0000393 -0.0029333 0.0000003 0.0071025 0.0000004
8 0.0006778 -0.0000172 0.0137509 -0.0000001 -0.0061440 -0.0000007
9 -0.0920592 -0.0000495 0.1713652 -0.0000002 0.1966698 0.0000001
10 0.0479442 -0.0000921 -0.2138562 0.0000011 -0.3091815 0.0000010
11 0.0792852 0.0000021 -0.1099086 -0.0000016 -0.2832366 -0.0000012
12 0.0847900 -0.0000838 0.0756669 0.0000016 0.4260712 0.0000012
13 0.1143711 -0.0000630 0.0601549 -0.0000016 -0.0078605 -0.0000012
14 -0.3333546 -0.0000690 0.0723164 0.0000004 -0.1231947 0.0000003
15 -0.3538504 -0.0001026 0.0357654 -0.0000006 0.0578532 -0.0000003
16 -0.0706189 -0.0000726 0.1495864 0.0000003 0.0956946 -0.0000001
ITER = 1201
1 0.0174145 15.4142496 -0.0001410-25.6926112 -0.0004992-10.4557301
2 0.0273388 35.8094305 0.0015937 -3.2579433 0.0011953 12.8007666
3 0.0086421 13.8197901 -0.0014862 6.2366361 -0.0011423-13.8626416
4 0.0227827 2.6600268 0.0015420-13.3679105 0.0012041 -7.5291023
5 0.0204389 45.1578993 -0.0014389 24.1375954 -0.0010458-28.7982789
6 -0.0064369-58.4173726 0.0004090-16.2926925 0.0004428 10.0752261
7 0.0407059-33.9341150 -0.0002803 -3.0413834 -0.0004000 7.3642022
8 0.0178253 0.7028110 0.0001337 14.2574699 0.0006960 -6.3703908
9 0.0512889-95.4505841 0.0002579177.6782010 -0.0001327203.9149797
10 0.0954598 49.7104663 -0.0011652*********** -0.0010502***********
11 -0.0021812 82.2060486 0.0016153*********** 0.0012471***********
12 0.0868566 87.9136330 -0.0016180 78.4544408 -0.0012298441.7673733
13 0.0653423118.5844534 0.0016355 62.3709488 0.0012653 -8.1500766
14 0.0715845*********** -0.0003655 74.9805224 -0.0002602***********
15 0.1064114*********** 0.0006686 37.0829400 0.0003627 59.9844847
16 0.0752908-73.2204880 -0.0003076155.0970508 0.0001312 99.2199353
ITER = 1241
1*********** 18.0560001*********** -0.1461529*********** -0.5175839
2*********** 28.3459128*********** 1.6524539*********** 1.2393501
3*********** 8.9604446*********** -1.5409031*********** -1.1843817
4*********** 23.6220140*********** 1.5987917*********** 1.2484695
5*********** 21.1918162*********** -1.4918756*********** -1.0843687
6*********** -6.6740595*********** 0.4240615*********** 0.4590700
7*********** 42.2054647*********** -0.2905768*********** -0.4147510
8*********** 18.4819802*********** 0.1386035*********** 0.7216104
9*********** 53.1783773*********** 0.2674182*********** -0.1375733
10*********** 98.9764982*********** -1.2080968*********** -1.0888817
11*********** -2.2615914*********** 1.6747825*********** 1.2930771
12*********** 90.0563561*********** -1.6775860*********** -1.2751234
13*********** 67.7494583*********** 1.6957174*********** 1.3119595
14*********** 74.2216035*********** -0.3789887*********** -0.2698126
15***********110.3314991*********** 0.6931838*********** 0.3760115
16*********** 78.0644970*********** -0.3189491*********** 0.1360462
ITER = 1281
1**********************151.5370417***********536.6512811***********
2******************************************************************
3******************************************************************
4******************************************************************
5******************************************************************
6******************************************************************
7**********************301.2814365***********430.0301406***********
8******************************************************************
9********************************************142.6413696***********
10******************************************************************
11******************************************************************
12******************************************************************
13******************************************************************
14**********************392.9503741***********279.7522466***********
15******************************************************************
16**********************330.6989797*********************************
ITER = 1321
1******************************************************************
2******************************************************************
3******************************************************************
4******************************************************************
5******************************************************************
6******************************************************************
7******************************************************************
8******************************************************************
9******************************************************************
10******************************************************************
11******************************************************************
12******************************************************************
13******************************************************************
14******************************************************************
15******************************************************************
16******************************************************************
(相変わらず先入観に因われる)
筆者は最初、上記の挙動を非常に不思議なものと思っていました。原子位置
は理想位置で力はゼロになるはずで、結果もそうあるべきと思い込み、実は
(初期化していないという)単純な問題であることに気付けませんでした(何
かもっと深刻な問題ではないかと考えた)。
(に)本日のバグ(レポート5/31、2013)
[目次]
本日のバグは、単純なスペルミス(typo error)に相当するものです。内容は、
GR(1)とすべきところを、GR(I)としていました。DO I=1,Nループ内の変数
GR(I)をそのままコピー&ペースト(書き写した)した後、I → 1に直して
いませんでした。"GR(1)"なので、結果への影響は非常に軽微ですが、現在計
算結果の検証を行なっています(5/31、2013)。←検証の結果、影響
は全く無視出来る(問題ない)ことが判明しました(6/6、2013)。
本バグは、コードの変更作業中に、たまたま当該部分を見ていたら、違和感
がありよくよく見ていたら、GR(1)であるべきところが、GR(I)となっていまし
た。この間違いは、ほとんど当該コード開発段階からそのままで、これまで計
算してきたことになります。ただ影響はほとんどないはずで、計算条件によっ
ては全く影響が出ない場合もあります。むしろ、問題はプログラム(コード)
の修正で、筆者はバージョン管理ソフトを使用していないので、"I → 1"の修
正もいちいち全てのプログラム上で編集しないといけない状況にあります。
間違いに気付かなかった(見過ごしてきた)のは、"I"と"1"の形が似ていた
こともあります。"K"とか"IJK"とかだったら、この間違い(誤り)は起こらな
かった(或いは、もっと早い段階で気付けた)かもしれません。
その当該部分を以下に示します。
SIGSTR(6)=SIGSTR(6)+STMQ+SS1*GZ(1)*GZ(I)+SSZ
"I"の部分を色分けしているため、上記では容易に見分けがつきますが、実
際の色分けされてないコードで、これを目視で(先入観なしに)見つけ出すの
は難しいと思います(実際、筆者は何年も気付かなかった)。
(は)本日のバグ(レポート3/22、2013)
[目次]
今回実は、引数不一致、配列未定義(或いは、未設定、値代入忘れ等々)、
配列の定義範囲の不一致からくる異常動作などを扱うことを企図していたので
すが、十分な結果が集められませんでした。そうこうしている内に新たな失敗
に遭遇しました。このため今回はこの新手の失敗について話したいと思います。
この新たな失敗は、座標入力のミス(誤り)です。それも複数の誤った座標
を使って計算を行なってしまいました。まず第一番目の誤りは格子定数の設定
です。
あるスーパーセルの格子定数の初期
値を以下のように設定しました。
50.0000000000 0.0000000000 0.0000000000
0.0000000000 5.3050000000 0.0000000000
0.0000000000 0.0000000000 5.3050000000
本当の妥当な値は以下の通りです。
32.0000000000 0.0000000000 0.0000000000
0.0000000000 5.6570000000 0.0000000000
0.0000000000 0.0000000000 5.6570000000
特に最初の値は、1.5倍以上大きな値(50 a.u.、1 a.u. = 0.529177 Å)と
してしまいました。より大きな値に設定したため、その分必要な計算量がかか
り、大変な無駄となってしまいました。これに気付いたのは、構造最適化を行
なっていくと、格子がどんどん小さくなり、50 a.u.から32 a.u.辺りまで小さ
くなっていきました。あまりに値の変化が大きくおかしいと思い、格子に関す
る検算を行なってみると、32 a.u.辺りが大体正しい値であることが分かりま
した。実際、構造最適化を進めると、この値近辺で収束します。
次に犯していた間違いは、格子(スーパーセル内の原子)の座標です。最初に設定した座標は、
0.0000000000 0.0000000000 0.0000000000
0.1250000000 0.0000000000 0.0000000000
0.2500000000 0.0000000000 0.0000000000
0.3750000000 0.0000000000 0.0000000000
0.5000000000 0.0000000000 0.0000000000
0.6250000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
0.8750000000 0.0000000000 0.0000000000
0.0000000000 0.5000000000 0.5000000000
0.1250000000 0.5000000000 0.5000000000
0.2500000000 0.5000000000 0.5000000000
0.3750000000 0.5000000000 0.5000000000
0.5000000000 0.5000000000 0.5000000000
0.6250000000 0.5000000000 0.5000000000
0.7500000000 0.5000000000 0.5000000000
0.8750000000 0.5000000000 0.5000000000
でした。まずこれが間違いであると気付き、修正を以下のように行ないまし
た。
0.0000000000 0.0000000000 0.0000000000
0.1250000000 0.0000000000 0.0000000000
0.2500000000 0.0000000000 0.0000000000
0.3750000000 0.0000000000 0.0000000000
0.5000000000 0.5000000000 0.5000000000
0.6250000000 0.5000000000 0.5000000000
0.7500000000 0.5000000000 0.5000000000
0.8750000000 0.5000000000 0.5000000000
0.0000000000 0.5000000000 0.5000000000
0.1250000000 0.5000000000 0.5000000000
0.2500000000 0.5000000000 0.5000000000
0.3750000000 0.5000000000 0.5000000000
0.5000000000 0.0000000000 0.0000000000
0.6250000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
0.8750000000 0.0000000000 0.0000000000
これで正しいと筆者は思っていたのですが、これも誤りであることが判明し
ます。計算の収束が遅く、どうもおかしいと思っていて、”ふと”おかしいこ
とに気付きました。そして最終的に正しい座標は、以下のようになります。
0.0000000000 0.0000000000 0.0000000000
0.1250000000 0.5000000000 0.5000000000
0.2500000000 0.0000000000 0.0000000000
0.3750000000 0.5000000000 0.5000000000
0.5000000000 0.0000000000 0.0000000000
0.6250000000 0.5000000000 0.5000000000
0.7500000000 0.0000000000 0.0000000000
0.8750000000 0.5000000000 0.5000000000
0.0000000000 0.5000000000 0.5000000000
0.1250000000 0.0000000000 0.0000000000
0.2500000000 0.5000000000 0.5000000000
0.3750000000 0.0000000000 0.0000000000
0.5000000000 0.5000000000 0.5000000000
0.6250000000 0.0000000000 0.0000000000
0.7500000000 0.5000000000 0.5000000000
0.8750000000 0.0000000000 0.0000000000
これが正しい座標です。この座標を使うと、ずっと計算の収束が速くなりま
した。この過ちは、既に述べたように、ある時、ふっと気が付きました。気付
いていなかったら、多分どこかの段階で必ず気付ける内容と筆者は思いますが、
より大変なことになっていた可能性があります。そもそも最初の座標構築と設
定の段階で、もう少し慎重であるべきだったと言えます。検算(検証)は、忘
れずにかつ可能な限り頻繁に行なうことが吉(必須)と言えます。
(別の誤りに気付く)
上記とは別件ですが、些細な間違いがありました。それは必要な数より多く
の数で配列を定義していた(数の不一致)ことによる間違いでした。これによっ
て結果に影響はありませでした。当該する数は、k点の数で4点で計算が足り
るところで、16点分を定義していました。通常、この手の定義では不足して
いる場合と違って、より多めに設定しても計算に問題ないようになっている場
合が多いです(実際の計算では必要な数までしか計算しないように設定してあ
ることが多いため)。ただk点に関してはそうなっていない部分があり、より
多めに定義した部分の計算を行なおうとしたため計算途上でエラー表示が出て
きました(表示に関わらず、計算は正しく遂行された。←5点目以降はエラー
で計算しないようになっていたため)。エラー表示部分近辺の数値の値を調べ
た結果、原因(数の不一致)を突き止めました(3/27、2013)。
PARAMETER(NK=16) ← 本当は、NK=4だった
DIMENSION A(NK)
DO 20 K=1,NK
CALL DIAGONAL(A,IERR) ← K=5以上は、エラー表示して計算しない
WRITE (6,*) 'IERR = 300 : ',IERR
20 CONTINUE
(更に別件のバグを発見)
またしてもバグを発見しました。気付けたのは、新しい系で計算してみると、
明らかに値があるはずの結果がゼロになっていたのを発見したためでした。当
該する計算部分を調べてみると、やはりコード上に誤りがありました。誤りは、
配列内の変数、AZ(N,L)とすべきところを、AZ(N,3)と定数(N → 3)にしてい
ました。また更に周りを調べると、BZとあるべき変数が、BYとなっていました
(Y,Zは座標軸方向を意味する)。おそらくコードを作成する段階で、コピー
アンドペーストをした部分を修正せずにそのままにした(例えば、Y方向部分
を扱う行をZ方向用にコピーして、Y→Zとし忘れた)ため、バグとして残った
ものと思われます。
(4/25、2013、追加)コピーアンドペーストでの間違え例として、
配列内の変数の間違いを最近また犯しました。A(I,NNN)を別の場所にコピーし
た時、その場所のDOループ内の変数が、NNNではなくIKなのに、そのままで計
算しておかしな結果を返していました(←実行途中でエラーで止まる。そのエ
ラーメッセージから問題点が見つかりました)。ただ、そもそもの話として、
同じ変数なのにNNNとかIKとかDOループ毎に異なる変数名にしていることの方
が問題ではあります。
(誤りは続く:今回は”である調”でいく。5/16、2013)何か悲し
いが、失敗は尽きない。今度の失敗は、事前に導出しておいた表式通りにプロ
グラム(コード)を組んでいなかった。表式通りでないことに、全く気付かず、
いろいろと計算を進めていた。問題が発覚したのは、表式の確認の過程で改め
てコードとの対応を調べ直したら、表式に対応するはずのコード(プログラム)
部分が、そうなっていなかった。
具体的には、積分を∫dr3とすべきところを、∫dr2
で計算していたり、表式では乗じていた、|k+G|(k点+逆格子ベクトルのノ
ルム)を実際の計算では、し忘れていたりした。
(ろ)本日のバグ(レポート6/26、2012)
[目次]
今回はバグと言うより、入力数値の設定ミスです(やはりバグと言えるかも
知れない)。以下は、過去にも提示したデータの一部ですが、最初の行の最初
の数値、"1000"は、プログラム実行におけるイタレーションの最大数を指定し
ています。筆者のプログラムでは、修正最急降下法(MSD法)を採用しているので、構造
最適化を含めた全イタレーション数は割と多いです。計算資源の量と計算資源
(計算機)の速さ、プログラムの収束の速さ(扱う系や計算条件等に依存する)
などとの兼ね合いから、大体1000回程度以内と指定していました。
1000 0.9500 0.1000D-07 6.5000 0
65.8112000000 0.0000000000 0.0000000000
0.0000000000 5.8169400000 0.0000000000
0.0000000000 0.0000000000 5.8169400000
0 COORDINATES 0:NORMALIZED 1:CARTESIAN
0.0061633466 0.0000000000 0.0000000000
0.0663263417 0.5000000000 0.5000000000
0.1273274737 0.0000000000 0.0000000000
0.1885776509 0.5000000000 0.5000000000
しかし実際の計算では、全イタレーション数が1000回を越えることもし
ばしばあります。上記の、数値"1000"は最大数として指定されますが、プログ
ラム実行における実際のイタレーション数の指定は別の部分でも行なっていま
す(←この同じようなものを2重で設定することは良くない)。で、これまで
の計算では、偶然この問題は顕在化しませんでした(しないような計算しかし
ていなかった)。今回たまたま問題が顕在化するような設定条件になり、計算
結果がおかしな結果を与えました。
それは、上記のデータのままにイタレーションの最大数を1000回と指定
した上で、実際の計算は1600回イタレーションを繰り返すものでした。で、
問題は電荷混合の比を格納する配列では、1000回分までしか指定しないよ
うになっていたことです(上記例示した数値データを使って設定するようになっ
ていた)。つまり1001回目以降は、未定義となり電荷混合が正しく行なわ
れません。このため1001回目から全エネルギーの値が、全く収束せず、振
動する事態となりました。
ここで、電荷密度の混合比(単純混合)を格納しているルーチン
(サブルーチンINPUT1)を示します。ルーチンの最後の方で、
PMIX(I)=PPMIXと混合比の値を格納しています。で、ITEMAXが上記データで指
定しているイタレーションの最大数。今回、実際のイタレーション数は、
ITEMAXより大きくなっていました。
これまでこのような設定になることがしばしばあったのですが、今回と少し
計算設定が異なるため問題が起こらないようになっていました(それで気付く
のが遅れた)。今回の計算では、その問題を回避する設定にならずに顕在化し
た訳です。
(い)本日のバグ(レポート10/13、2011)
[目次]
今回もまたやってしまいました。今回の失敗も大変単純でした。
ある並列計算(OpenMPを使用)で、力の値がおかしいことに気付きました。
計算対象の構造の持つ対称性から、そもそも力がゼロになるべきなのに、有限
の値が得られていました。それも同じ条件で計算すると、出てくる結果がまち
まちで、たまに力がゼロ(正しい結果)になることもありました(計算毎に値
が違う)。全エネルギーなどの値は、どの場合でも正しい結果を与えました。
明らかに、並列計算関連でおかしなことになっていると分かる状況ですが、原
因究明に割と時間がかかりました。
原因は、力の計算部分での足し上げのところでした。逐次計算なら問題ない
のですが、並列で計算する場合に何も対処していないとおかしな結果となる部
分がありました。対処の仕方は、REDUCTION(OpenMP)によって変数を指定し
ておくことです。
!$OMP DO REDUCTION(+:ZFORC2)
ZFORC2が足し上げ対象となる変数。この1行がなかったために、毎回計算す
るたびに異なった結果(力の値)を与えていました。
そしてもっと問題なのが、この失敗は既に過去に経験していたことです
(参考文献参照)。並列化されたプログラムのバージョン(版)管理にも
問題がありました。筆者は、いわゆるバージョン管理ソフト等を使用していま
せん。で、今回、古いバージョン(版)のプログラムで並列計算を行なってし
まいました。筆者は、そのことに全く気付きませんでした。本当は、大元の最
新プログラムと比較検討することが容易に可能であり、そうすべきだったので
すが、全くそれを行ないませんでした。
REDUCTIONは、並列計算で数値の足し上げを各CPU毎に行なう時、それぞ
れのCPU上で計算された値をまとめあげるための指示を与えます。そうしな
いと正しい足し上げ結果とならずに、実行毎に値が異なったりもする。ただ、
資料によってはREDUCTIONで指定する変数はスカラー変数のみで配列変数は駄
目という記述も散見されます。筆者の場合、REDUCTIONで指定している変数
(ZFORC2)は配列変数(並列計算に対しては独立)です。一応、それでも正しく
動いていますが(非並列時の結果と整合している)、これに関しては更に検討・
調査を行なっています(調査に結果、新しいバージョン〔FORTRAN〕では配列
変数の指定が可能な模様)。
関連ページ:OpenMP導入に関するメモ
のメモ1参照。
参考文献:小林一昭、「徹底解剖 第一原理計算:第九回”計算
上の落し穴(問題点)”」、金属、第80巻、第6号(2010年6月号)、
58頁(日本語)、[情
報](J-GLOBAL, jst)
問題は、これまでも何度も指摘したように、同じか或いは類似した失敗を繰
り返していることである。これにはより根源的(研究姿勢、環境等)な問題、
欠陥の存在が示唆される(のかもしれない)。
(Z)本日のバグ(レポート1/21、2011)
[目次]
今回の問題は、バグとは言えないものですが、ここに記しておきます。ある
バンド計算プログラムを動かしている時、これまで問題なく動いていたものが、
新たな系を扱ったところ突然計算途中で止まってしまうようになりました。計
算が突然停止したことに対する直接的なエラーメッセージはなく、流したジョ
ブのエラーメッセージも(筆者にとっては)意味不明なものでした。最初は、
単純な入力データの間違いと思い、いろいろ調べましたがどのようにしても計
算が途中で止まってしまいました。このバンド計算プログラムの場合、対称性
の設定や、単純に単位胞内の原子数の設定を間違っても、計算が(何のエラー
メッセージなく)止まってしまうのですが、今回はそれとは異なりました。入
力等の設定間違いの場合、ジョブ全体に関してのエラー表示はないのですが、
今回は意味不明ながらエラー表示(メッセージ)がありました。
ちょっと悩んだ末に、原因はメモリが足りないということに気付きました。
案の定、同じ系で計算規模を少なくすると(今回の場合、k点数を少なくして
みた)、計算は完結するようになりました。メモリー不足の場合、これまでは
意味のあるエラー表示(見てすぐに判断出来るということ)が出ていたのです
が、今回は少なくとも筆者には全く意味の分からない表示(メッセージ)しか
ありませんでした。このため原因に気付くのが遅れました。
今回使用したプログラムは、メモリを浪費する版だったので、少し大きな系
を実行させようとすると障害(今回のように突然止まる)が起きることは、十
分に気を付けていればすぐに「メモリ周りが怪しい。」と気付いたところです
が、そうなりませんでした(やはりエラー表示が理解出来なかったのが痛かっ
た。が、これを理解出来る方が筆者はおかしいと思う。残念ながら当該表示
〔メッセージ〕は、ここで明らかにすることが出来ません)。
尚、当該プログラムは、よりメモリを消費しない版に改良し、当初扱った系
でも問題無く動く(計算が完結する)ようになりました。
(Y)本日のバグ(レポート3/2、2010)[目次]
今回も盛大にやってしまいました。非常に単純ながら大きな失敗をしてしま
いました。
今回の失敗はプログラムのバグではありません。実は、計算としては間違っ
た計算ではなく正しい計算を行なっています。しかし大変な失敗計算になって
います。
で、どのような間違いを犯したかと言うと、ある系のバンド計算を行なう時、
原子数60個/単位胞、1個当りの電子数は4個(価電子数)という前提で、
241バンドとして計算してしまったことです。ここでスピンは縮退
して(非磁性の場合)います。241としたのは、240バンドまで電子が詰
まっているとして(←ここ重要)、1個分多くしました。扱った系はギャップ
が空いていて、それもワイドギャップなので1バンド分多いだけで問題ありま
せん。少なくともより小さい系での検証では、全く問題ありませんでした。
しかし、これは大変勘違いな計算をしていることになります。電子数240
個の系で、スピンが縮退している場合、必要なバンド数は120です。
実際は、金属である場合もあるので、これより多めのバンド数で設定します。
広いギャップが空いている系なら、多めと言ってもほんの少しで十分です。1
21でも大丈夫なはずです(バンドが非常に狭い場合などは注意が必要)。実
際は、バンド数、123で計算(241としていた場合と比べて同じ全エネル
ギーを返すことを確認)しています。
バンド数を241として計算しても間違いではありません。この場合系は金
属ではなく、バンドギャップを持っているので、241バンドの内121バン
ド分が空のバンドとして計算されています。これらの空のバンドは、実質計算
結果に寄与しません。空のバンドからの寄与が必要な、GW近似などでは重要ですが、今回の計算は
ごく普通のバンド計算(ギャップの空いた系の基底状態を求める電子状態計算)
なので空のバンドの影響はありません。つまり正しい結果を返している訳です。
その意味では間違いではない。
しかしながら、これは原子数60個という筆者がこれまで扱ってきた系でも、
最も大きな部類に入ります。勿論、昨今のバンド計算ではもっとずっと大きな
系の計算も平気で行なわれていたりします。しかし、それでも120バンド程
度で済む計算を、240バンド(正確には241バンド)で行なっていました。
バンド計算の計算時間は、大体バンド数に比例します(注:グラムシュミット
の直交化は、バンド数の2乗のオーダー)、メモリーもおよそ比例します。つ
まり2倍分の余計な計算をしてしまっていたのです。それもかなり時間が経つ
まで気付きませんでした(気付いたのも偶然に近い。ふと定義、設定を見てい
て気付いた)。これはちょっと悲しいです。
バンド計算を始めてから25年、未だにこのような初歩的なところで間違い
を犯しています。情けない限りです。
尚、120バンドまで電子が詰まっている系で、241バンド(121バン
ドは空のバンド)という、大量の非占有バンドがある状況の計算は、これまで
計算した経験がなかったが、今回試すことが出来た。計算を進めると全エネル
ギーは完全一致ではなくなったが、精度的には依然問題ないレベルの差しかな
い。
(3/5、2010)この間違いに気付いたのは、既に述べたように本当に
偶然だった。ふとインクルードファイル”
PACVPP”(参考例、今回のものとは異なります)を見ていたら、「あれっ?」
と思い気付いた(その前に、別のプログラムソースを見ていて違和感を憶えた
のが始まり)。ちょうどその時、別件でソフトウェアの導入で苦労していて、
漸くうまくいきかけていた頃で、気分的(大変心許ない話だが)に気付き易い
状況にあったのかもしれない。
(X)本日のバグ(レポート9/4、2009)[目次]
今回の失敗は、プログラム上のバグではない。これとは別に、最近小さなレ
ベルでのバグの発見や、検証計算があった。例えば、未定義の配列を使用して
いて、通常は動いていたが、ある条件の計算で突然実行が止まって(←当該部
分に、IF判定によるSTOP命令があった)、気付くということがあった。
で、今回の間違いは、形成エネルギー計算における、単純で深刻な誤りだっ
た。形成エネルギーに関しては、形
成エネルギーと凝集エネルギーの関係を参照して欲しい。計算で、化合物
の各構成元素1原子当たりのエネルギーに換算するためのユニットセル内の原
子の数が間違っていた。少ない数で割ったため、化合物のある構成元素の原子
1個当たりのエネルギーを過剰に低く(安定に)評価したため、得られる形成
エネルギーがかなり損になる(不安定な)値になってしまった。
あまりにべらぼうに損(不安定)になる値であったので、再度検討をしてみ
た結果、ユニットセルの原子数の値が間違っていることに気付いた。ちょっと
情けない話である。検算はちゃんと行なわなければならない。
(追記:12/4、2009)
今回追加として扱うべき誤りがありました。基本的に今回の誤りは、過去の”
(B)本日のバグ(レポート11/18、1998)”
と同じものです。入力データとしての擬ポテンシャルデータに間違いがありま
した。元素A、B、Cがあったとして、ABCという順で擬ポテンシャルデータを作
るべきところを、ABDと全く関係ない元素Dの擬ポテンシャルが入ったデータを
入力データとして使ってしまいました。他の関連入力データ、設定は、ABCと
いう前提で計算したので、これでは全く正しい計算になっていません。原因は、
確実に詰めた訳ではありませんが、元々ACDという入力データがあり、これか
らABCを作成しようと、エディタ上で編集した際に間違いが起きたと考えられ
ます。ACDからDを削除後、ACにBをAとCの間に挿入するのが正しい作業手順で
した。おそらく、ACDにBを、ABCDとなるように挿入し、削除すべきはDなのにC
を削除し、ABDを作ってしまったようです。
残念ながら、それでも筆者のプログラムは正しく動いてしまいます。正しく
動作しますが、当然結果は正しくありません。この間違いに気付いたのは、全
エネルギーから形成エネルギーを求めると、目茶苦茶不安定になる結果が出た
ためです。通常は、不安定になると言っても、数eV/原子以下のエネルギー差
ですが、数a.u.(ハートリー原子単位)/系全体の差(原子当りでも、数十eV
以上)が出ました。いくら何でもこいつはおかしいと思って、データを調べて
いたら先の擬ポテンシャル並びに関する誤りに気付きました。
(W)本日のバグ(レポート8/5、2008)[目次]
今回のバグは、計算した系の総電子数にゴミが出るというものです。それは、
サブルーチンXCFFT
で総電子数(ユニットセル内の系の電子の総数)を計算すると、以下のような
値(総電子数80個の場合)になることから発覚しました。
REAL TOTAL CHARGE = 80.0026633146275827 IN XCFFT
本当の総電子数は80個/ユニットセルですが何故か、0.0026633146275827
とゴミのような余計な部分が出てきます。もし正しく総電子数が求まるなら、
以下のような値になります。
REAL TOTAL CHARGE = 80.0000000000007 IN XCFFT
ゴミ(誤差)部分は無視出来るほど小さな値になります。この問題は、これ
までとは異なる数値演算ライブラリのFFTルーチンを利用した場合に起こりま
した。当初は、これに全く気付きませんでした。それは総電子数にゴミがある
のに、全エネルギーなど他の値に問題が生じなかった(総電子数が正しく出る
場合と値が完全に一致する)ためです。こんなに電子数の値に違いがあるのに、
何で全エネルギー等が一致するのが最初不思議でした(後述。←現在でも完全
に裏付けは取れていない)。で、結局原因と思われる部分として以下のルーチ
ンが浮かび上がりました。このルーチンの一番外側のIF文の中にあるIF文、
"IF ( MOD(I,IFX2).NE.0 ) THEN"ですが、これはFFTにおけるメッシュに関係
するもので、筆者が元々使用していたFTTルーチン(MFFT)では、3次元複素フー
リエ変換において、メッシュ数:IFX2が2n-1でした(nは適当な正の整数)。
FFTとして使用するメッシュは、2n-2個分必要で、それより一つメッシュが多
い仕様でした。従って、2n+1番目のFFT用の配列は、実際の物理量の計算には
関わらない部分なので、総電子数計算の際に相当する部分の電荷密度の値
(CHGB1(I ← IFX2))をゼロとしました。ただ、以下のルーチンでは問題があ
ります。一番外側のIF判定は、CHGB1がゼロ以下の場合という制限を与えてい
ます。従って、そうでない場合、次のIF判定である、"IF ( MOD(I,IFX2).NE.0
) THEN"が意味を持ちません。
MFFT使用時は、以下のような判定処理をしないでも正しい値が得られるよう
になっていました。あくまで、2n+1番目の部分は作業用でしか使用されないし、
それが結果に影響を与えないようになっていました。今回は、MFFTではなく他
のFFTルーチン(数値演算ライブラリ)を使用しました。そもそも今回のFFTで
は、2n+1番目の部分は必要なかったのですが、元々のプログラムがMFFTで動く
仕様になっていあたため、それと整合させるため、2n+1(実際使用されるのは、
2n-2まで)として計算していました。で、筆者は以下のルーチンで、2n+1部分
の処理は行なわれていると思っていました。前述のように、2n+1番目に相当す
るCHGB1が必ずゼロ以下である必然性はなく、実際2n+1番目にゴミ(としての
数値)が残ったままで計算が行なわれてしまった訳です。そしてこれが結果と
して総電子数のゴミとして現れました。
ICCCC=0
DO 20 I=1,KSUM
IF( CHGB1(I).LE.0.0D0 ) THEN
IF ( MOD(I,IFX2).NE.0 ) THEN
IF ( CHGB1(I).LE.-1.0D-5 ) THEN
WRITE(6,610) I,CHGB1(I)
610 FORMAT(1H ,'**WARNING CHG.DEN<0.0 AT ',
& I5,2D15.7,'***')
ICCCC=ICCCC+1
END IF
CHGB1(I) = 1.D-40
ELSE
CHGB1(I) = 1.D-40
END IF
END IF
20 CONTINUE
S = 0.D0
DO 1010 I=1,KSUM
S = S+CHGB1(I)
1010 CONTINUE
S=S*VCEL - SCHGPC
IF (MOD(ITER,10).EQ.0) THEN
WRITE (6,*) 'REAL TOTAL CHARGE = ',S,' IN XCFFT'
END IF
この問題の解決は、以下のようにルーチンを変更することによって達成され
ます。
ICCCC=0
DO 20 I=1,KSUM
IF( CHGB1(I).LE.0.0D0 ) THEN
IF ( CHGB1(I).LE.-1.0D-5 ) THEN
WRITE(6,610) I,CHGB1(I)
610 FORMAT(1H ,'**WARNING CHG.DEN<0.0 AT ',
& I5,2D15.7,'***')
ICCCC=ICCCC+1
END IF
CHGB1(I) = 1.D-40
END IF
IF ( MOD(I,IFX2).EQ.0 ) THEN
CHGB1(I) = 1.D-40
END IF
20 CONTINUE
S = 0.D0
DO 1010 I=1,KSUM
S = S+CHGB1(I)
1010 CONTINUE
S=S*VCEL - SCHGPC
IF (MOD(ITER,10).EQ.0) THEN
WRITE (6,*) 'REAL TOTAL CHARGE = ',S,' IN XCFFT'
END IF
以上にあるように、2n+1であることを判定するIF文(深紅色部分)を独立さ
せてやれば、総電子数の値に現れるゴミ数値の問題は起こらなくなります。
最後に、今回のバグでは総電子数に問題がありましたが、全エネルギー等に
問題はありませんでした。おそらくこれは、総電子数は逆格子空間の電荷密度
ρ(G)を逆フーリエ変換(Inverse FFT = IFFT)して、ρ(G) → IFFT → ρ(r)
を求めました。ρ(r)は実空間での電荷密度(バンド指標、k点指標などは省略)。
全エネルギー等の値は、畳み込みなど
の過程を経た項が元となっています。この場合、先の逆フーリエ変換したもの
を更にフーリエ変換して元の逆格子空間に戻しています。これにより影響を与
えた2n-1部分(実空間)が、再び2n-1部分のみに集約(逆空間)されて結果に
影響を与えなかったのではないかと考えています。ここら辺は、完全には確か
め切れていません(↓追記も参照↓)。
(2/9、2009追記)変数SCHGPCにも問題があることが判明しました。
SCHGPCは、部分内殻補正を使用する場合に必要となる変数ですが、これの初期
化は部分内殻補正をするサブルーチン内のみで行なわれていました。部分内殻
補正をしない場合でも上記ルーチンにあるように、総電荷数の足し上げの時に
使用されています(この時初期化されていない)。状況によってはこの時、
SCHGPCに自動で初期化されず、不定の値が収まってしまう可能性があることが
判明しました。プログラムの最初で、SCHGPC = 0.0D0と初期化して置くと、こ
の問題は解消されます。これは同じプログラムでも、システムによって問題無
かったり、少しプログラムが異なるだけで現れなかったりすることが分かって
います(調査中)。ただ、SCHGPCのような変数は必ず初期化して置くことで、
常に使用するのにある条件の時だけ初期化(或いは数値の代入)が行なわれる
ようにしないことです。
(V)本日のバグ(レポート2/8、2008)[目次]
また間違った擬ポテンシャルを読み込んで計算していました。これは、これ
までのバグレポート(J)、(B)
と同じ失敗です。元素としては同じものながら、価電子の扱いが異なる擬ポテ
ンシャルを誤って用いていました。これは比較的浅い内殻の電子を価電子とし
て扱うものと、扱わない擬ポテンシャルがあり、内殻を考慮しない計算の場合
に、内殻を価電子として考慮した擬ポテンシャルを内殻非考慮と思い込んで使
用していました。全く過去の教訓が生かされていません。そもそも相変わらず、
このような場合の誤りを事前、或いは少なくとも計算実行初期に確認(チェッ
ク)する機能が導入されていません。情けない話です。
(U)本日のバグ(レポート12/17、2006)
[目次]
今回のバグは、非常に単純なものです。バグ(と言うか問題)のあったプロ
グラムは、バンド計算プログラム本体ではなく、バンド計算結果を使って解析
を行なうごく簡単なプログラ
ム(バンドギャップのある系の、直接、間接を判定する)です。このプロ
グラム自身は一応(?)問題無いのですが、重大な制限があります。それはユ
ニットセル内の総電子数が16個(h-BNはボロン、窒素が各2個ずつで構成さ
れる)でないと正しく動作しません。勿論、系が半導体(絶縁体、つまりギャッ
プが存在する系)であることも必要です。またスピンは縮退しているとして、
1バンドに2個電子が詰まることを想定しています。
ところが筆者はこれを、ボロン、窒素がユニットセルに各4個ずつ存在する
系(総電子数32個=16バンド)で、そのまま使用してしまいました。プロ
グラムを見れば分かるように、この場合完全に詰まっている8番目と9番目の
バンドが計算の対象となり、これによって得られた判定結果は全く意味のない
ものになります。バンドギャップの値も負の値という変なものになってしまい
ます(←本当はここで最低限、エラーを返す〔警告する〕ようにしなければな
らない)。最初、筆者はこの問題に気付かず、暫く経ってから結果のおかしな
ところ(先の負のバンドギャップ値)に気付き、そこからプログラムの不備に
辿り着きました。対策は、応急的ですが判定すべきバンドを8番目、9番目か
ら、16番目(VBMがあるバンド)、17番目(CBMがあるバンド)に変
えることで正しい結果が出ました。VBM:価電子帯の頂上。CBM:伝導帯
の底。
この問題はプログラムを最初に作った段階で認識していたのですが、将来へ
の対策を全く施して(考えて)いませんでした。で、案の定、問題を忘れてそ
のままでは計算していけない系に適用し、挙げ句に間違いに気付くのに暫く時
間を要するという失敗の連鎖を行なってしまいました。
(12/27、2006)間違いは別にまだあった。電子数が増える方では
なく、減る方もあった。つまり、ユニットセル内の全電子数が16→8に減る
場合もあった。この時、系がギャップを持つなら、電子に占有されたバンド数
は4であり、非占有の一番下のバンド(空のバンド)は下から5番目というこ
とになります。筆者は、これも見逃して占有8バンド、最低非占有9バンド目
として計算していました。勿論、正しい結果は得られていませんでした(バン
ドギャップも負の値という変な値となっていた。←ここで気付くべきだった)。
(T)本日のバグ(レポート8/21、2006)
[目次]
今回のバグは既に過去に言及したものと同じものです。そのバグは、(K)です。古いバージョンのプログラムを動かしたところ、
計算されたストレスの値に、同じ条件での計算結果にかかわらず一致しない場
合が出てきました。最初、原因不明でしたが、新しいバージョンのプログラム
と、問題のる当該プログラムの差分を取り、詳しく調べたところ問題点が判明
しました。その内容は(K)でのものと全く同じでした。つまり、サブルーチ
ンFORZFBがストレス計算に対応していませんでした。これを修正することによ
り問題は解決しました(←完全に一致することを確認)。
プログラムのバージョン管理をちゃんと行なう必要があります。古いバージョ
ンのプログラムでも、時としてそれを使用して計算結果を得なければならない
事態もあり得るので、古いバージョンのプログラムに対する、バグ等への対応
を怠らずに行なう必要があります。
いろいろな事情で、古いバージョンのプログラムを使用せざるを得ない状況
は存在し、当該プログラムより後のバージョンでは対応済みのバグが、そのプ
ログラムに残っている可能性があります。そのような場合の対応は、使用者、
作成者本人もそのことを忘れる、或いは認識していない可能性もあり、悩まし
い問題の一つです。出来るだけ、バグの記録を詳細に残し、どのバージョン、
どの(バージョンの)プログラムまで対応したか、きちんと管理する必要があ
ります(まあ大抵それが出来ていないことが多い)。
(S)本日のバグ(レポート9/16、2005)
[目次]
本日の失敗(→教訓)は、結晶対称性に関してのものです。今回扱った系は、
C6B2系が主なものです。これは仮想物質なのですが、
元々計算を始めた、Na3As構造のC6B2以外
の仮想構造の計算で問題が発生しました。C6B2
(Na3As構造)については、第17回化合物新磁性材料研究会[講演
]参照。
C6B2(Na3As構造)の結晶対称性は、
P63/mmcとhcp構造と同じ高い対称性を持ちます。しかし、この構
造は完全な仮想構造で、(凝集)エネルギー的にも安定でないことが計算によ
り分かっています。そこで、これと類似の仮想構造の候補を考え、それらの安
定構造と電子状態を求めることとしました。
最近のコードは大変良く出来ていて、与えた系(ここでは
C6B2系仮想物質)の結晶対称性を自動でも求めてくれ
たりします。これまで扱っていた、C6B2
(Na3As構造)と類似の新たな仮想C6B2に
ついて、その結晶対称性を自動判定したところ、その結晶対称性では筆者のプ
ログラムで計算出来ない(結晶対称性の判定テスト部分で止まる)ことが判明
しました。当初、この原因が全く分からず、いろいろなことを試しましたが全
てうまく行きませんでした。
C6B2(Na3As構造)の原子座標は、筆者
の計算プログラムでは、
25.9099482945 0.0000000000 0.0000000000
0.0000000000 4.7796946433 0.0000000000
0.0000000000 2.3898473216 4.1393369833
0 COORDINATES 0:NORMALIZED 1:CARTESIAN
0.2500000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
0.0000557124 0.3333333333 0.3333333333
-0.0000557124 0.6666666667 0.6666666667
0.4999442876 0.3333333333 0.3333333333
0.5000557124 0.6666666667 0.6666666667
0.2500000000 0.3333333333 0.3333333333
0.7500000000 0.6666666667 0.6666666667
となります。上記は既に構造最適化を終了しています。" 0 COORDINATES
0:NORMALIZED 1:CARTESIAN "の行以降が原子座標で、最初の6行が炭素(6個)、
最後の7、8行がホウ素(2個)です(以下同じ)。" 0 COORDINATES
0:NORMALIZED 1:CARTESIAN "行の上にある数値は、格子座標です。座標は、筆
者の計算では(z,x,y)となっています。従って、一番左側の数値がc軸(=z軸)
のものです。また、"0.6666666667 0.6666666667"というような表現も一般的
でありません。通常は、"0.6666666667 0.3333333333"などと表現します。
まず以下の仮想構造での原子座標、
0.2500000000 0.0000000000 0.0000000000
0.2500000000 0.3333333333 0.3333333333
0.7500000000 0.0000000000 0.0000000000
0.7500000000 0.6666666667 0.6666666667
0.0000000000 0.3333333333 0.3333333333
0.5000000000 0.3333333333 0.3333333333
0.0000000000 0.6666666667 0.6666666667
0.5000000000 0.6666666667 0.6666666667
を考え、あるコード(←筆者のものでない)で結晶対称性を判定をしたとこ
ろ、187番(P6m2←本当は数字6の上にバーが付く)という解答でした。と
ころが、筆者のバンド計算プログラムコードで187番の結晶対称性として計
算させようとすると、結晶対称性判定部分で止まってしまいました。筆者のコー
ドは結晶対称性の処理に不完全なところがあり、他の対称性では上記座標でも
計算が通ってしまうのですが、この場合、計算結果が明らかにおかしなものと
なります。この問題に際して、当初は結晶対称性に関してのオペレーター(演
算子)の設定が筆者のコード上で間違っているのではないかと、検証、検討を
試みました。とにかく、判定を通るように強引な設定で計算を走らせてみたり
とか、187番の対称性は、P6/mmm対称性からインバージョン(反転)を除い
たものなので、P6/mmmで強引に計算させてみたりしましたが、どれもうまく行
きませんでした。
そこで今度は、同僚の新井さんに相談、別の計算コードで結晶対称性の判定
を行なってもらいました。筆者は、この構造は187番の対称性ではないので
はとも考えていたのですが、このコードでも判定結果は187番でした、ただ
原子座標の設定(原点の取り方)がちょっと上記のものと異なるという結果が
出てきました。つまり以下のような座標の移動(変換)が必要であることが分
かりました。
(1) 元の原子座標
0.2500000000 0.0000000000 0.0000000000
0.2500000000 0.3333333333 0.3333333333
0.7500000000 0.0000000000 0.0000000000
0.7500000000 0.6666666667 0.6666666667
0.0000000000 0.3333333333 0.3333333333
0.5000000000 0.3333333333 0.3333333333
0.0000000000 0.6666666667 0.6666666667
0.5000000000 0.6666666667 0.6666666667
(2) x,y座標を、0.3333333333だけずらす。
0.2500000000 0.3333333333 0.3333333333
0.2500000000 0.6666666667 0.6666666667
0.7500000000 0.3333333333 0.3333333333
0.7500000000 0.0000000000 0.0000000000
0.0000000000 0.6666666667 0.6666666667
0.5000000000 0.6666666667 0.6666666667
0.0000000000 0.0000000000 0.0000000000
0.5000000000 0.0000000000 0.0000000000
(3) z座標を、0.25だけずらす。
0.5000000000 0.3333333333 0.3333333333
0.5000000000 0.6666666667 0.6666666667
0.0000000000 0.3333333333 0.3333333333
0.0000000000 0.0000000000 0.0000000000
0.2500000000 0.6666666667 0.6666666667
0.7500000000 0.6666666667 0.6666666667
0.2500000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
(4) 単なる並べ替え。
0.0000000000 0.3333333333 0.3333333333
0.0000000000 0.0000000000 0.0000000000
0.5000000000 0.3333333333 0.3333333333
0.5000000000 0.6666666667 0.6666666667
0.2500000000 0.6666666667 0.6666666667
0.7500000000 0.6666666667 0.6666666667
0.2500000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
(5) 単なる並べ替え。
0.0000000000 0.0000000000 0.0000000000
0.0000000000 0.3333333333 0.3333333333
0.5000000000 0.3333333333 0.3333333333
0.5000000000 0.6666666667 0.6666666667
0.2500000000 0.6666666667 0.6666666667
0.7500000000 0.6666666667 0.6666666667
0.2500000000 0.0000000000 0.0000000000
0.7500000000 0.0000000000 0.0000000000
上記の最後の原子座標と、最初の原子座標は等価なものです。ただ、最後の
もの(5)では、座標(0,0,0)に炭素原子が置かれているようになっています。対
称性に関してのオペレーターは、対象となる原子座標の配置による影響を受け
るため、正しい配置にしておかないと、判定に失敗したり、正しい計算が出来
ないということがこれで分かりました(←もっと早く認識すべきだった)。
上記最後の原子座標は、新井さんが使用したコード上で示された(変換され
た)座標で、筆者のコードでもこの座標では、187番の結晶対称性で正しく
計算を遂行することが出来ました。最初に筆者の使用したコード(←筆者独自
のコードでない)では、おそらく座標の変換を内部で自動的に行なっているも
のと推定されます。コードに慣れていないため、その過程を出力結果から見い
出せていない可能性があります。
今回、異なるコード(手法、手段)を使用した結果
の比較によって、問題を解決することが出来ました。
【参考】対称性、群論関連の[サイト]
(R)本日のバグ(レポート2/11、2005)
[目次]
本日の失敗は、ごくごく単純です。しかし、だからこそ重要。六方晶構造の
単位胞での基本格子ベクトルを与える時、a,b,c各軸とそれらの各軸同士がな
す各度α、β、γにおいてa,b軸のなす各度(γ)を、筆者は60度としてい
ました。ここではc軸をz方向とし、a軸、c軸及びb軸、c軸のなす各度はいずれ
も90度とします。あるソフトで、γを60度として、六方晶格子の格子デー
タを入力して計算させると、どうも計算結果がおかしな値になり、困っていま
した。教科書(結晶格子について載っている固体物理関係のもの)をちゃんと
見ると、γは120度となっていました。で、γを120度として当該ソフト
で計算すると、確かに妥当な結果が得られました。
この場合、γを60度として格子を作って話を進めても、それ自身は間違い
ではないです。少なくとも、それに対応するようにソフトウェアコードが動く
ようにさせていれば問題ありません。自作のものならそれでOKなのですが、
一般のもの(フリーソフトなり市販品なり)ではそうとも行きません。γを6
0度とするのは一般的ではなくほとんどの場合、慣例としてγは120度とし
て格子パラメータは設定されます。筆者はこのことを十分認識していませんで
した(←筆者の勉強不足が問題)。
因みに、このソフトはWyckoffパラメーターを入力すれば良いようになって
います。この時、筆者は初めて、Wyckoffパラメータの存在と意味を認識しま
した。結晶構造データベース[Crystal Lattice Structures Database](←現在、
当該ページは存在しない。The Naval Research Laboratory)には、ちゃんと例
示されている結晶構造のWyckoffパラメーターが表示してあります。筆者はこ
のサイトを大変重用していて改めて良くできていると思いました。
(Q)本日のバグ(レポート9/26、2005)
[目次]
何と、本ページ自身に失敗があった。(Q)レポートを飛ばして(P)の次
を(R)としていた。アルファベットの順番のチェックを怠っていた。悲しい。
(P)本日のバグ(レポート1/12、2005)
[目次]
今回のバグ(と言うより失敗)は2つあります。一つはバンド計算時の座標
入力上のもので、今一つは擬ポテンシャル作成時の入力データ間違い(ミス)
です。いずれも比較的(または非常に)単純なものす。1/12以降、大変多
忙な日が続き、実際のアップロードは、2/7(2005)となりました。
(1)座標データの間違い。これは六方晶構造の物質の計算において、ユニッ
トセルではなく、スーパーセルとして
計算しようとした時に問題が発生しました。ユニットセルの規模の計算では、
その系の持つ対称性から、ユニットセル内の原子が受ける力は(多くの場合)
ゼロでした。←六方晶と言っても、ウルツ鉱型構造のように内部パラメーター
を持つ場合、原子が力を受ける場合があります。今回の場合、ユニットセルの
規模では原子は力を受けないような系だったのですが、それをスーパーセルと
して計算を行なうと、想定していない力を原子が受ける事態に遭遇しました。
これはユニットセルとして計算できる系をユニットセル数個分(例:1×1→
2×2)のスーパーセルと単純に拡張しただけで、スーパーセル内の原子はユ
ニットセルでの場合と同様、原子は力受けないはずにも関わらず、実際の計算
では力が出てきてしまいました。
当初、筆者は、このようなことはキュービック(立方体。或はもっと緩めに
四角→直方体)の場合ではこのような力が出てくることはなかったので、六方
晶に特有の問題(例:k点の問題とか)と全く根拠のない推定のもとに問題を
放っていました。しかし、それは全くの事実誤認でした。最近、プログラムの
出力に関して若干の改良を施し、それによるテスト計算を上記のスーパーセル
からなる系に対し行なってみた結果、明らかにおかしな結果が出てきました。
それは、改良した計算ではユニットセル(スーパーセル)内の各原子間の距離
を計算し、大きさ順に並べる結果を出力させるようにしたのですが、それが明
らかに予想した結果と異なるものでした。これは前述の力の問題以上に深刻で、
あり得ないことでした。これらの事実からより詳細な検討を行なった結果、筆
者のデータ入力の仕方が誤っていたことが判明しました。データの入力を正し
いものにすると、あり得ない力は消え、原子間距離などに対し妥当な結果が得
られました。今回、プログラム的には、変更箇所は全く無く、データの入力の
仕方を変えるだけで事足りるものでした。あり得ない
ことやおかしな結果が出てきたら、希
望的、楽観的な解釈(予想)から問題が解決した(している)と考えず、徹底
的に検討するべきだと改めて痛感しました。
(2)今一つの失敗は、擬ポテンシャル作成時のもので、明らかに存在しない
エネルギー準位を指定して擬ポテンシャル(Os)を作成していました。この場合、
Os(オスミウム)は、1s〜6pまで15の準位が存在するとして計算すべき
ものでした。ここで、6pは空の準位として考えます(←空にするのは、基底
状態のs、dを考える時で、pを扱う時は、6pをイオン化させて空でない準位
として扱う)。ところが実際は、この15番目の6pを16番目と指定して擬
ポテンシャル作成していました。この状態で作成したOsは、バンド構造として
はそう変なところはなかったのですが、実験値と比べて平衡格子定数が非常に
伸びた値になってしまいました。非常に長い間、この間違いに気付かなかった
のですが、ごく最近入力データを良く見ていたら15個までしか準位が存在し
ないのに16番目の準位が指定されている間違いに気付きました。
早速、間違いを修正、作成し直したOs擬ポテンシャルで、平衡格子定数を求
めると正しい妥当な結果が得られました(LDAによる計算ながら若干格子定数
は伸び目となる)。そもそもあり得ない準位を指定しても計算が通り、もっと
もらしい結果が得られたということも問題の一つであります。これについても
今後検討する必要があります。
(O)本日のバグ(レポート2/25、2004)
[目次]
久々のバグです。これはバンド計算でよく使用される高速フーリエ変換に関
するものです。最近は、LiBCや、
MgB2などのボライド系の計算が多く、あまり遷移金属の計算は行
なっていませんでした。そして、ある遷移金属の計算をしてみたところ、大変
計算が遅いことに気付きました。遷移金属の場合、3d、4dなどのd電子を価電
子として考慮する必要があり、その分計算が面倒になるのですが、今回の計算
は通常のバルク系で、hcp構造(つまりユニットセル内に原子は2個)が計算
対象であり、計算規模も先のLiBC、MgB2と比べて、ほとんど変わ
らないものでした。
しかしながら、実際に計算してみるとめったやたら計算速度が遅いことが判
明しました。遷移金属だから特殊な事情があるのかとも考えましたが、使用す
るプログラムは、LiBC、MgB2の時と同じもので、計算条件や設定
もさして変わらないので、そのような”特殊”な事情は存在しないと判断され
ます。
原因は気が付けば、今回も大変単純なものでした。単に、高速フーリエ変換
で使用するパラメーターの設定の仕方が妥当なものでなかったからでした。高
速フーリエ変換(FFT)では、主に電荷密度などに対して実空間⇔逆格子空
間の変換を行なうのですが、その時に使用する格子の数(空間のメッシュ数)
は、2の倍数である場合が最も高速となります。そうでない場合でも2、3、
5などからなる倍数で、17とか23のような値の倍数の選択は計算速度を非
常に遅くするので推奨されません。FFTのプログラムによっては2の倍数或
いは、これに準じる2、3、5の倍数でないものはそもそも計算できない設定
となっているものもあります。
今回筆者が使用したFFTルーチンは、2(2、3、5)以外でも計算可能
なもので、34(17×2)を使っていました。これはFFT計算部分を大変
遅くすることが判明しています。
(どうやって気付いたか)
たまたま、上記のFFTルーチンが使用できないマシン上で、昔から使って
いるMFFT(2、3、5の倍数しか使えないFFTルーチン)を使用したため、
34ではエラー表示が出たため気付きました。このことがなければ、今でも気
付かないままでした。通常は、メッシュ数が2、3、5程度の倍数でない場合
は、それに近いより大きい2、3、5位までの倍数になる値で設定していたの
ですが、今回はそのまま気付かずに34(17×2)のままで計算していまし
た。この計算では、結局32(2の5乗)の値も使えました(←本来なら36
だが、筆者は更に勘違いしていて32でよいものをわざわざ34で計算してい
た)。34と32でのバンド計算全体での速度差は、粗い評価ですが10倍以
上異なることが判明しています。
もう既に何度も書いていますが、計算がどこかおかしい時、たとえば(1)
計算結果は正しいが動作がおかしい(例:計算速度がやたら遅いなど)、(2)
動作速度などは問題ないが計算結果が微妙に(或いは何となく)おかしい、
(3)計算結果も動作もどこか(微妙に)おかしい、などの場合、計算に何か
問題があるはずで、放置せずに原因究明に努めることが必要と考えます。
(N)本日のバグ(レポート4/9、2003)[目次]
プログラムというものは、これまでやったことがない新しいことをしようと
すると、トラブルに見舞われることが良くあります(筆者の場合特に)。
今回のバグは、筆者の使っているバンド計算プログラムにおいて、新しい系
を計算した時に起こりました。勿論、プログラムは、この初めて扱う系にも仕
様上は正しく計算できるはずでした。
しかし、実際計算を行なうと、ストレスなどが正しく計算できていないこと
が判明しました。
では今回扱ったのはどのような系だったのか、物質名は具体的にはまだ挙げ
られないのですが(正直、そんなに秘密にするほどの系ではない)、ある3元
系で、アルカリ金属(擬ポテンシャルにおいて、s、pのみ非局所)、遷移金
属(s、p、dとも非局所)、酸素(s、pのみ非局所)からなる化合物(酸
化物)でした。内殻補正は、アルカリ金属と遷移金属の擬ポテンシャルに対し
て施されていました(今回は、これは影響なし)。
問題となったのは、非局所の部分に対応する、局所ポテンシャル部分の扱い
のところでした。これは、計算結果がおかしいことに気付いた後、検証のため、
元素の座標の入力の順序を、アルカリ金属、遷移金属、酸素の順番を、遷移金
属、アルカリ金属、酸素と変えると、計算結果のある部分に違いが存在するこ
とから判明しました(当然、本当は元素の座標の順序の入れ換えで結果に影響
はないはず)。違いがあったのは、全エネルギーの値が順序入れ換えで異なり、
更に細かく調べると、局所擬ポテンシャル部分に関わる値がおかいしことが判
明しました。以下が、当該する部分のプログラムルーチンです、
DO 500 II=1,KTYP
ETOT1 = ETOT1 + FLOAT(IATOM(II))*PAI*ACHG(II)
& * ( AC(II,1)/BC(II,1) + AC(II,2)/BC(II,2) )/UNIVOL
TOTCH=TOTCH + FLOAT(IATOM(II))*ACHG(II)
C
IF (ITER.EQ.0) THEN
IF (NLSPD(II).EQ.1) THEN
READ(16,*) MESHR,NMES,DX,RAD,VD,VDNL
DO 1210 N=1,MESHR
VDD(N,II)=VD(N)
VDDNL(N,II)=VDNL(N)
1210 CONTINUE
ELSE
DO 1212 N=1,MESHR
VDD(N,II) =0.0D0
VDDNL(N,II)=0.0D0
1212 CONTINUE
END IF
ELSE
DO 1211 N=1,MESHR
VD(N)=VDD(N,II)
VDNL(N)=VDDNL(N,II)
1211 CONTINUE
END IF
C
S=0.0D0
DO 1200 N=1,MESHR
S=S + OMO(N)*VDNL(N)*(RAD(N)**3)
1200 CONTINUE
色(深紅)のついた配列VDNLですが、実は
これが正しくゼロ初期化できないことがバグの原因でした。
3元系なので、DO 500ループ繰り返し数は3で、その都度、各元素に対応し
た局所ポテンシャル(VDNL)を読み込むのですが、VDNLは、s、p、dが非局
所の場合は読み込まずゼロ初期化するようにしています(IF文による分岐)。
しかし、実際に初期化しているのは、VDDNLで、VDNL自身ではありません。と
ころが、後の部分(S=S + OMO(N)*VDNL(N)*(RAD(N)**3))でVDNLが使われています。こ
の部分は、まだDO 500ループ内です。つまり、もし3元系の元素の順番が上記
アルカリ金属、遷移金属、酸素なら、最初、アルカリ金属(s、p非局所)で、
VDNLが読み込まれ、次の遷移金属の時は、VDNLはゼロとして扱われるべきです
が、上記ルーチンでは(上記で示していない部分を含めて)、VDNLは初期化さ
れておらず、前のアルカリ金属の値がそのまま残っています。このため、Sが
余計に計算され、局所部分の値がおかしくなります。
(解決法)
非常に簡単で、上記ループで、
DO 1212 N=1,MESHR
VDD(N,II) =0.0D0
VDDNL(N,II)=0.0D0
VDNL(N) =0.0D0
1212 CONTINUE
と初期化をきちんとすれば問題は解決します。
(何故、今まで気付かなかったか?)
例えば、LiBC(LiBCの研究ページ)や、
MgB2(MgB2の研究ペー
ジ)の計算を筆者は行なっていて、LiBCも同じ3元系で、上記と同様な問
題が生じてもおかしくなかったのですが、”幸運”にも、偶然このバグは発生
しないようになっていました。それは、LiBCでは、Liのみが、s、p、d非局
所であり、この最初の元素がs、p、d非局所なら、VDNLの読み込みは行なわ
れず、局所ポテンシャル部分の計算結果はおかしくなりませんでした
(MgB2も同様の理由)。
但し、これは、偶然筆者の使用するシステムが、暗黙の内に未代入、未使用
の配列の値をゼロ初期化しておいてくれたためで、計算システムの仕様によっ
ては、ゼロ初期化されずに、おかしな値となる可能性があります。
今回の教訓は、今までちゃんと正しく動いたプログラムでも、新しい系、条
件の計算では、いきなりおかしな結果となることがあること、おかしな結果を
与える原因(バグ)も決して複雑なものであるとは限らず、今回のように大変
単純なものであることの方が、むしろ多いということです。しかし、これによ
りプログラムはより強力(Robust)で信頼性の高いものになっていく訳です。
構成元素の原子座標の順番を変えることで、バグを発見できたのは、バグ発
見の手法、手順としては典型的と言えます(参考ペー
ジ)。
(M)本日のバグ(レポート12/5、2002)
[目次]
実に1年ぶりのレポートです(ここ最近あんましコードを書いていなかった)。
今回のバグ・失敗も大変単純なものでした。
(バグ・失敗詳細)
k点の座標を読み込むルーチンを作成して、そのk点座標を基にしてバンド
計算を行なうようにプログラムを変更し、実行させたのですが、テスト段階で
既に結果が分かっている系での、k点座標を読み込ませて計算した結果が、正
しい結果と一致しないという事態に遭遇しました。
(原因)
これは割と簡単に発見できました。k点座標を読み込むためのプログラムで
は、まず大元のデータから必要なk点座標を抽出し、それを掃き出すプログラ
ムを作り、その掃き出したデータを、実際にバンド計算プログラムから読み込
むようにさせました(この2段階の操作に問題があった←何でこのような面倒
なことになるかについては、いろいろ事情があったりする)。
筆者の扱うバンド計算プログラムは、ベクトルマシン用のチューンの影響が
まだ生きていて、なるべく3次元配列(例:A(1000、100、100))
の最初の次元での配列の数を大きめにとるようになっています。この影響で、
六方晶系を扱う場合、c軸(普通はz座標〔c軸方向の格子定数は大抵a,b軸よ
り長い〕で、3次元配列なら、最後の次元〔つまりz座標〕)をx座標として
扱うようにしていました。
このため第一段階のk点座標抽出・掃き出しプログラムにおいて、以下の様
に元データ中から座標を読む(READ文)段階で、z座標→x座標に変換するこ
ととしました(変換する手としては他にも沢山ある:
今回のREDA文での方法を推奨している訳ではない)。
READ(20,*) KVZ(NNN),KVY(NNN),KVX(NNN),KQWGT(NNN)
本来は、KVX(NNN),KVY(NNN),KVZ(NNN)というk点座標の並びを、READ文で読
み込む時の配列の並びを、KVZ(NNN),KVY(NNN),KVX(NNN)として、KVX(NNN)→
KVZ(NNN)、KVZ(NNN)→KVX(NNN)となるようにしました。
これ一回で座標の変換は終了なのですが、筆者はやってしまいました。この
掃き出したデータを、バンド計算プログラム側で読み込むルーチンのREAD文で
も、再び、
READ(61,*) KVZ(NNN),KVY(NNN),KVX(NNN),KQWGT(NNN)
としてしまいました。これは変換を2度行なっている(つまり、x→z→x)
ことであり、元に戻していることに他なりません。
このため計算結果が、一致しなかった(元の正しい結果は、上記第一段階で
の変換した座標での計算結果)のでした。上記のバンド計算側のREAD文を、
READ(61,*) KVX(NNN),KVY(NNN),KVZ(NNN),KQWGT(NNN)
とすると、比較すべき正しい結果と、計算結果の一致を見ることができまし
た。
(どうやって気付いたか)エラーの原因は、
バンド計算プログラム側で読み込んだk点座標をWRITE文で確認してみると、
最初の元データの座標と同じ並びに戻っていることから気付きました。
(補足)
尚、上記計算において、六方晶の格子座標の取り方の違いによる座標変換が
必要でした。上記バグとは関係ないのですが、参考までにその手続きを以下に
示します。
筆者の計算プログラムでは、実空間での六方晶の格子を、
13.1338124721 0.0000000000 0.0000000000
0.0000000000 5.1692358601 0.0000000000
0.0000000000 2.5846179299 4.4766895732
のように与えています。これに対応する逆空間での格子座標は、
0.478398 0.000000 0.000000
0.000000 1.215496 -0.701767
0.000000 0.000000 1.403534
です。バグの説明のところでも述べましたが、x座標がc軸となっています。
ここでy、z座標の取り方に、上記とは異なる取り方があります。それは実空間
格子座標では、
13.1338124721 0.0000000000 0.0000000000
0.0000000000 2.5846179299 -4.4766895732
0.0000000000 2.5846179299 4.4766895732
という取り方です。これに対応する逆空間格子座標は、
0.478398 0.000000 0.000000
0.000000 1.215496 -0.701767
0.000000 1.215496 0.701767
となります。基のk点の取り方が、整数表示になっていたので、上記逆空間
格子座標を使って、目的のk点座標を求めることが出来ます。以下に、座標を
変換している部分を示します。
C
READ(60,*) KVX(NNN),KVY(NNN),KVZ(NNN),KQWGT(NNN) ←読み込み(整数)
C
KQSUM = KQSUM + KQWGT(NNN)
C
VX(NNN) = DFLOAT(KVX(NNN))/24.0D0 ←実数化
VY(NNN) = DFLOAT(KVY(NNN))/24.0D0
VZ(NNN) = DFLOAT(KVZ(NNN))/24.0D0
QWGT(NNN) = DFLOAT(KQWGT(NNN))/DFLOAT(KKX*KKY*KKZ)
C
C R22 = RLTV(2,2)
C R23 = RLTV(2,3)
C R32 = RLTV(3,2)
C R33 = RLTV(3,3)
R22 = RLTV(2,2) ←座標変換1
R23 = RLTV(2,2)
R32 = RLTV(3,2)
R33 = -RLTV(3,2)
WRITE (6,*) "R22 = ",RLTV(2,2)
WRITE (6,*) "R23 = ",RLTV(2,3)
WRITE (6,*) "R32 = ",RLTV(3,2)
WRITE (6,*) "R33 = ",RLTV(3,3)
C
Q(1) = VX(NNN) ←座標変換2
Q(2) = VY(NNN)
Q(3) = VZ(NNN)
QX=RLTV(1,1)*Q(1)+RLTV(1,2)*Q(2)+RLTV(1,3)*Q(3)
QY=RLTV(2,1)*Q(1)+R22*Q(2)+R23*Q(3)
QZ=RLTV(3,1)*Q(1)+R32*Q(2)+R33*Q(3)
VX(NNN) = QX
VY(NNN) = QY
VZ(NNN) = QZ
C
QSUM = QSUM + QWGT(NNN)
C
C WRITE(6,*) KNUM,VX(NNN),VY(NNN),VZ(NNN),QWGT(NNN)
C WRITE(6,402) KNUM+1,VX(NNN),VY(NNN),VZ(NNN),QWGT(NNN)
WRITE(6,*) KVX(NNN),KVY(NNN),KVZ(NNN),KQWGT(NNN)
WRITE(6,402) KNUM,VX(NNN),VY(NNN),VZ(NNN),QWGT(NNN)
402 FORMAT((I4,4(F15.9,2X)))
400 CONTINUE
上記ルーチンは、あまりというかほとんど洗練されていません(2/13、
2003)。
(L)本日のバグ(レポート12/19、2001)
[目次]
久々に間違ってしまいました。相変わらず今回のバグも単純なものです。
プログラムに新しい対称性(Wurtzite型構造)に対応するよう改良を施す過
程で生じた間違いです。筆者の計算では、対称性は次のような番号で指定され
ていました。
C-----KBZTYP=1 : WHOLE B.Z. , 2 : SIMPLE CUBIC IR.B.Z.
C----- 3 : BCC IR.B.Z. , 4 : FCC IR.B.Z.
C----- 5 : DIAMOND STRUCTURE IR.B.Z.
C----- 6 : HEXAGONAL STRUCTURE IR.B.Z.
C----- 7 : P6/MMM(ALB2) STRUCTURE IR.B.Z.
C----- 8 : TETRAGONAL STRUCTURE IR.B.Z FOR BETA-TIN
C----- 9 : A-PBO2 STRUCTURE IR.B.Z FOR SIO2
C----- 10 : TETRAGONAL STRUCTURE IR.B.Z FOR RUTILE
C----- 11 : ORTHOROMBIC, CACL2 TYPE
C----- 12 : FCO IR.B.Z.
C----- 13 : PERPVSKITE(ORTHO) IR.B.Z.
これに新たに、Wurtzite型構造(六方晶型、P63mc)を加えるに
あたってちょっと困りました。もう既に0以上の番号(番号0は対称性を考慮し
ない表面系用)が埋まっていて、HCP構造のところに、この新たな対称性のた
めの番号を配することができなかったのです。そこで仕方なく以下のような番
号付けをしました。
C-----KBZTYP=1 : WHOLE B.Z. , 2 : SIMPLE CUBIC IR.B.Z.
C----- 3 : BCC IR.B.Z. , 4 : FCC IR.B.Z.
C----- 5 : DIAMOND STRUCTURE IR.B.Z.
C----- 6 : HEXAGONAL STRUCTURE IR.B.Z.
C----- -6 : WURTZITE STRUCTURE IR.B.Z.
C----- 7 : P6/MMM(ALB2) STRUCTURE IR.B.Z.
C----- 8 : TETRAGONAL STRUCTURE IR.B.Z FOR BETA-TIN
C----- 9 : A-PBO2 STRUCTURE IR.B.Z FOR SIO2
C----- 10 : TETRAGONAL STRUCTURE IR.B.Z FOR RUTILE
C----- 11 : ORTHOROMBIC, CACL2 TYPE
C----- 12 : FCO IR.B.Z.
C----- 13 : PERPVSKITE(ORTHO) IR.B.Z.
何と、”-6”という負の番号を強引に定義したのです。間違い(バグ)の原
因は、番号が負の整数値だったことです。プログラム内で、IF判定で各対称性
に対応した計算を行なっている時、これまでは対称性を考慮しない場合(番号、
0,1)と、対称性を考慮する場合(番号2以上)というようになっていたので、
IF判定で、IF (KBZTYP.GE.2)或いは、IF (KBZTYP.LE.1)という部分がプログラ
ム内に何箇所かありました。筆者は、この部分への対応を忘れてしまったので
す。
また、この番号を担うプログラム内の変数が、”KBZTYP”だけでなく、もう
一つ別に、”NBZTYP”というものがあり、筆者はこの”NBZTYP”に関する部分
の変更にのみ注目してしまったことが、”KBZTYP”絡みの部分への注意を失わ
せることとなりました。
当然の如く、計算結果に問題が生じました。計算して得られる力や、ストレ
スの値で、明らかに対称性からゼロにならねばならないものに有限の(無視で
きない)値が出てきてしまったのです。そして、その原因が暫くの間全く分か
りませんでした。
いろいろ試行している間、与えた結晶内原子の座標の設定に問題があるので
はとも考えたのですが、このWurtzite型構造による系を対称性を考慮しない条
件で計算した場合では、正しく力、ストレスの値が求まる(この場合の”正し
い”は、ゼロになるところは、ちゃんとゼロになるという意味)ことが判明し、
結晶内の原子位置の設定の問題ではないということとなりました。
バグを見つけた発端は、対称性が正しく計算されている場合と今回の計算の
結果との比較からでした。それは、対称性が正しく計算されている場合のスト
レスの結果、
TOTAL SUMM 1 = -3.391163378869085E-004
2 = 1.273323054860103E-003
3 = 1.716355131468628E-004
4 = -4.301059916385391E-004
5 = 1.596389500226893E-004
6 = 4.345718045303151E-004
TOTAL SUMM OP1 = -3.391163378869083E-004
2 = 1.129377263005734E-021
3 = -1.411721578757167E-020
4 = 2.232906445887955E-006
5 = -1.129377263005734E-021
6 = 2.232906445888011E-006
と、今回の場合の同様の結果、
TOTAL SUMM 1 = 1.523900247301507E-004
2 = 1.641143043416956E-005
3 = 1.089318435141048E-005
4 = 2.612009135650878E-004
5 = 1.295694267740399E-004
6 = 2.076013932175158E-004
との比較でした。見てお分かりの通り、後者では、”TOTAL SUMM OP”の計
算結果部分が抜けています。この最後の部分の結果は、それまで得られたスト
レスの値に対称オペレーターをかけて、対称性をきちんと考慮した結果を与え
るもので、この結果の部分が表示されていないということは、最後に行なわれ
る対称オペレーターをかける計算が行なわれていないことを意味します。
調べてみると、当該部分の対称性考慮、非考慮の判定が、”IF
(NBZTYP.LE.1) GO TO 9000”となっていました。これはこれまでの計算なら、
番号0,1の場合(対称性を考慮しない)は、対称オペレータをかけずに、文番
号9000に飛ぶというのでよかったのですが、今回のWurtzite型構造に対応
する番号は、-6で、これもこのままでは対称性を考慮しない計算として話が進
んで(文番号9000に飛んで)しまっていた訳です。前述のように、変数”
KBZTYP”に関しての判定部分のチェックを全く見過ごしていたことが間違いの
もとでありました。他の箇所も含め、変数”KBZTYP”の関わる部分(電荷密度、
力の計算部分)の修正を行なうことで、力、ストレスとも妥当な結果を与える
ようになりました。
今回の問題は、
(1)事前のプログラム設計の不備。番号が割り付けられず、負の整数値を導
入したところで駄目駄目と言えます。
(2)NBZTYP、KBZTYPという事実上同じ機能を持った変数が独立に存在して、
プログラム内で使用されている(プログラム改良時に一方が忘れられる可能性
があり、バグの元になる)。
と言えます。
(K)本日のバグ(レポート7/16、2001)
[目次]
今回のは単純で大きな間違い(バグ)でありました。
問題はストレスの計算に関しての部分で、症状は基本的に同じ計算を行なっ
ているはずなのに計算値が異なるというものです。まず、その結果を以下に示
してみます。
(ケース1)
TOTAL SUMM OP1 = 8.363462325643757E-004 <-- ここ
2 = 1.129377263005734E-021
3 = -1.129377263005734E-021
4 = -5.152418845876215E-004 <-- ここ
5 = 1.044673968280304E-020
6 = -5.152418845876215E-004 <-- ここ
(中略)
TOTAL ENERGY FOR 401-TH ITERATION= -51.5028059 -0.5150281D+02
MIXING RATE = 0.500000000000000
ITER=401 ET(H)= 0.1493166D-06 ET(M)= 0.004061 DC= 0.3162807D-06
(ケース2)
TOTAL SUMM OP1 = -3.459023787458713E-003 <-- ここ
2 = 0.000000000000000E+000
3 = 1.807003620809174E-020
4 = 1.641161644709054E-003 <-- ここ
5 = -2.258754526011467E-020
6 = 1.641161644709055E-003 <-- ここ
(中略)
TOTAL ENERGY FOR 401-TH ITERATION= -51.5028059 -0.5150281D+02
MIXING RATE = 0.500000000000000
ITER=401 ET(H)= 0.1493166D-06 ET(M)= 0.004061 DC= 0.3162807D-06
ケース1とケース2で、”ここ”で示した値(ユニットセルにかかる圧力
〔ストレス〕)の値が異なっています。一方、全エネルギー(TOTAL ENERGY)
の値は一致しています。上記には示されていませんが、力の値も一致します。
値が異なるのはストレス部分のみです。上記2つの場合での計算条件の違いは、
ケース1では電子状態のみの計算を行ない、最後にストレスの値を計算し、そ
れを表示させる。ケース2は、セルに関しての構造最適化を行なう過程で、最
初に電子状態計算を行なわせ、セルの構造最適化を始める直前でのストレスの
値を計算表示させるものでした。いずれも上記の場合では、401回目(イタ
レーション)での値です。
この状況から見ると、原因追求は簡単かとも思えたのですが、バグ発見当日
には原因を突き止められませんでした。
状況として、(1)値が異なるのはストレスのみ、(2)全エネルギーなど
他の値に違いが無い、(3)計算条件における違いは、セルに関しての構造最
適化をさせるか、させないかだけの差、などが挙げられます。特に(3)から、
バグが潜んでいそうなのは、計算条件に関わる部分の可能性が高く、比較のた
めの(小規模な)テスト計算を繰り返したのですが、ストレスに関しての値が
異なることがますます明白になる一方で、どうして値が異なってしまうのかさっ
ぱり分からないという状態が翌々日まで続きました。
原因究明のきっかけとなったのは、ケース1、ケース2の計算出力結果を、
diffにかけて違いを比較検討したことでした。このこと自体は、バグ発見当日
にも行なったのですが、その時点では問題点を見い出せませんでした。翌々日
になって改めて見直してみると、違いが存在することに気付きました。それは、
以下の部分です。
3c3
< 0.500000000000000 0.000000000000000E+000 0.000000000000000E+000
---
> 0.500000000000000 0.000000000000000E+000 10.0000000000000
15c15
< 400 400 400 1 401 0
---
> 400 2000 40 1 2000 0
45,46c45,46
< IOVE = 400
< IMDI = 400
---
> IOVE = 40
> IMDI = 2000
50,51c50,51
< ISTOP = 401
< ISTMD = 401
---
> ISTOP = 2000
> ISTMD = 2000
72c72
< DTIMUC = 0.000000000000000E+000
---
> DTIMUC = 10.0000000000000
2855c2855
< CALL FORCE
---
> CALL FORZFB
2901,2906c2901,2906 (↓ストレスの計算途中の値)
< SIGNL 4 1 = -0.275069507467294
< 2 = 1.189155365786973E-003
< 3 = 2.122295586945811E-004
< 4 = -0.274503255358969
< 5 = 1.073767340287859E-003
< 6 = -0.275494497853064
---
> SIGNL 4 1 = -0.279364877487318
> 2 = 3.965559338907192E-003
> 3 = 2.529826782671025E-003
> 4 = -0.268815066930155
> 5 = -3.305458596529087E-004
> 6 = -0.276869879223285
(以下略)
上記(diffコマンドによる差の表示結果)で、(主に前半部分の)計算条件
の違いによる違いは別として、二つのことが指摘できます。
(い)”CALL FORCE”と”CALL FORZFB”の違い。(ろ)ストレス計算途中
の値の違い。まず、(い)からサブルーチンの呼び出しの仕方が異なることが
判明します。(ろ)に関しては、diffをかける前から、ストレスの計算途中の
各段階での値のどの部分に違いがあるか調べた段階でこのことは判明はしてい
ました。ストレスに関しては、その計算途中の値全てが異なっていた訳ではな
く、ある特定のルーチンでの結果(値)がおかしいことは分かっていたのです
が、どうしてこれが間違いに繋がるのか不明でした。それを解決に導いたのが、
(い)のサブルーチン呼び出しの違いでした。
サブルーチンFORCE
は、非局所擬ポテンシャル部分の計算を行なうルーチンで、このルーチンでは
原子に働く力において、非局所擬ポテンシャル部分が寄与する部分の計算を行
なっています。一方、サブルーチンFORZFB
は、サブルーチンFORCEで行なう計算の内、力に関しての部分を除いた計算を
行ないます。これは電子状態のみを求める計算では、力の計算が必要ではない
ため、この場合、より簡便で計算時間を消費しないルーチンに切替えて計算し
た方が良いためです。
ここで問題だったのが、このサブルーチンFORZFBは”電子状態”計算のみ用
でしかなく、”電子状態+ストレス”(原子は動かないとする場合)用になっ
ていませんでした。以下が、サブルーチンFORZFBの該当部分です。
DO 1000 IA=1,KATM
IF (NLSPD(KFTYPE(IA)).EQ.1) THEN
LNUM = 4
ELSE
LNUM = 9
END IF
DO 1003 L=1,LNUM
!XOCL SPREAD NOBARRIER DO /IP
DO 1001 IK=1,KV3
DO 1002 IBAN=NBD1,NBD2
ZFC(IBAN,IK,IA,L) =DCMPLX(0.0D0,0.0D0)
1002 CONTINUE
1001 CONTINUE
!XOCL END SPREAD
1003 CONTINUE
1000 CONTINUE
C!XOCL END PARALLEL
C VPP-PARALLEL START
C!XOCL PARALLEL REGION
!XOCL SPREAD NOBARRIER DO /IP
DO 2100 IK=1,KV3
DO 2110 IA=1,KATM
CS=1.0D0/(WS(KFTYPE(IA))*UNIVOL)
CP=1.0D0/(WP(KFTYPE(IA))*UNIVOL)
CWL(1)=CS
CWL(2)=CP
CWL(3)=CP
CWL(4)=CP
C
IF (NLSPD(KFTYPE(IA)).EQ.1) THEN
LNUM = 4
ELSE
LNUM = 9
CD=1.0D0/(WD(KFTYPE(IA))*UNIVOL)
CWL(5)=CD
CWL(6)=CD
CWL(7)=CD
CWL(8)=CD
CWL(9)=CD
(正)CWL(10)=CDを付加
END IF
DO 3222 L=1,LNUM
DO 3200 IBAN=NBD1,NBD2
C 1991 11/28 I ---> I1
DO 3510 I=1,IBA(IK)
I1 = NBASE(I,IK)
L1 = IG1(I1)+KX1
L2 = IG2(I1)+KY1
L3 = IG3(I1)+KZ1
ZTMP=ZZZ(I,IBAN,IK)*DCONJG( ZFM2( I1 ,IA ) )
ZFC(IBAN,IK,IA,L)=ZFC(IBAN,IK,IA,L)+
& ZTMP*SSS(I,IK,KFTYPE(IA),L)
3510 CONTINUE
ZFC(IBAN,IK,IA,L)=CWL(L)*ZFC(IBAN,IK,IA,L)
3200 CONTINUE
3222 CONTINUE
C
2110 CONTINUE
2100 CONTINUE
配列SSS()が非局所擬ポテンシャル項に関するもので、通常の電子状態の計
算では、l=2(d軌道)まで計算する場合、s、p、d合わせて9つ(LNUM
= 9)までの計算が必要となります。実は、本プログラムではストレスの計算
でのd軌道部分からの寄与として、LNUM = 10に相当する部分の計算が必要で
した(深紅色で示した部分)。これを忘れた
ためストレス部分の計算結果が一致しない事態となった訳です。勿論、LNUM =
10として計算をし直すと、結果は一致するようになりました。
(最近行なった、MgB2の計算に関して)
一軸圧縮による、MgB2の計算を最近行なって、学会発表や、投
稿を行なっていたのですが、上記問題の影響はあるかどうかについて言及する
必要があります。
結論から言うと、確かにこの影響はあったが、不幸中の幸いにも最終結果や
結論に影響を与えるほどの誤差(0.1 GPaのオーダー以下)になりませんでし
た。MgB2においては、このLNUM = 10に相当する部分の寄与が結果
として小さかったのが幸いしました。また、電子状態のみの計算(サブルーチ
ンFORCEでは、LNUM = 10に相当する計算は正しく行なっていた)でのチェック
計算でも、MgB2は十分正しく収束(含むストレス)していること
が確認できていました。
今回、問題に気付いたのは、MgB2以外に、最近他の理論計算の
予測(プレプリントサーバーで発見)で、CuB2、AgB2、
AuB2などが高い超伝導転移温度を示すというものがあったため、
筆者もこの系の計算を試みてみようと思い、平衡格子定数をセルの最適化から
求めようとしたら、予想と大分異なった値になったことでした。いろいろチェッ
クする内に今回のバグにまで行き当たった次第です。実際、当該ルーチン部分
をコードしてから数年経つのに、これまで全く気付かなかったのが問題です
(あまりストレス計算を必要とする機会がなかったというのもあるが)。
今回の教訓としては、これまでと異なる未経験の系を計算してみておかしな
結果が出てきた時は、プログラムのバグも疑えということです(常にバグであ
るという訳ではない。←実はこれも問題:バグかバグでないかを確認するのも
困難が伴うことが多い)。
(厳密に言うと)
上記の修正は、ストレスの計算を行なわないで、電子状態だけを計算するだ
けなら、LNUM = 10の部分だけ余計に計算することになるので、若干無駄な計
算をしていることになります。ただ、今回のバグの遠因として、いろいろな計
算条件毎に計算を場合分けすることにより、計算が複雑化し、それがバグ発生
の要因となったとも言えます。従って、今回は上記処方で、”よし”としまし
た(勿論、もっとエレガントな方法もあるかと思うが、、、)。
(更にお恥ずかしい話)
何と、このバグは既に報告([バグレポート3])され
ていた!。筆者に学習能力や、プログラム管理能力が無いことが歴然!。
ただ、現在と過去とで、本バグとその対策(対処法)に対する認識の違いは
興味深い。
(2/17、2012)あまり使用していない古い版(だが、その版でしか
出来ない計算)のプログラムで再び同じ過ちを犯してしまう(と言うか、ちゃ
んと版管理をすべきであり、そうしておけばこのような間違いの繰り返し、再
生産は生じませんでした)。悲しい。
(2/21、2012)上記と関連して、更に不十分な点がありました。従
来のプログラムの結果と全エネルギーが一致しないことから問題が発覚(本来
なら値がおかしいのはストレス値で、全エネルギーの値は正しい)しました。
この時、計算条件によっては、全エネルギーの値が正しくなるのが曲者(プロ
グラムとしては正しくないが、偶然うまく動いてしまう)。不一致の原因は、
配列定義が、修正部分とそれ以外の従来部分とで矛盾するためでした(本来、
A(N,M,L)とすべきとろこを、A(N,M)としていました。L = 1のみの計算だとう
まくいってしまう。←状況に依る)。このため当初、この問題に気付けません
でした(2/22、2012、文章若干修正)。
(J)本日のバグ(レポート3/2、2001)[目次]
新世紀発の失敗です。今回は、現時点で原因の不明な錯誤によるものです。
今回のは、プログラムのバグではなく、入力としてのデータの間違いによる
ものでした。問題は二つあり、現行の筆者のシステム(プログラム実行に至る、
過程として)では、今回の失敗を完全に回避することができないことと、長期
間この失敗に気付けなかったことです。
失敗は、バンド計算で間違った擬ポテンシャルのデータを入力として使い続
けてしまったことです。まず、バルクの計算を行ない、平衡格子定数、バンド
構造等を求めました。この段階で、この物質は仮想的な物質だったため、他に
比較すべき実験値、理論値の資料がありませんでした。バンド構造に、特に問
題はなかったのですが、全エネルギーの出方にはちょっと変なところがありま
した。
それは、平衡格子定数を求めるため、格子定数-全エネルギーの計算を行なっ
ていると、全エネルギーの最小値近傍で、全エネルギーの値がふらつくことで
した。つまり、非常に細かく格子定数を変化させると、全エネルギーが、大き
くなったり、小さくなったりで、ぎざぎざとなることです。これは、格子定数
の変化による平面波数の急な変化によるものではないです(この場合の格子定
数の変化よりずっと小さい)。また他の系では、このようなことはほとんどあ
りませんでした。
で、この時点では、格子定数の変化(0.002 a.u.)が小さいものによるもの
(誤差)で、単に他の系より全エネルギーが敏感なだけと思い、そのまま計算
を次の段階(バルクから表面系へ)に進めてしまいました。
ちょっとまだ曇行きが怪しくなってきているので、現在記述を凍結中です
(7/16、2001)。
(I)本日のバグ(レポート9/18、2000)
[目次][関連]
今回の過ちは、前回のバグ[レポート]と深く関連して
います。
(9/19、2000)表面の双極子モーメントを求めるために、筆者は3
次元の実空間に関しての積分を行なっていました。これに必要な実空間電荷密
度は、フーリエ変換(注:高速ではない)により求めていました。従って、そ
れに要する計算時間は、4重のループ(実空間用に3重、逆空間用に1重)に
なり、大変膨大なものになります。Alphaチップマシン(21264使用)でさえ、
計算に2、3日もかかってしまいました。これは甚だ効率の悪いものであると
言えます。
偶然、数年前に書かれたコードサンプル(筆者のものではない)を見ている
内に、この実空間での積分とは異なる手法が存在することに気が付きました。
それは、実空間での電子の電荷密度に関する双極子モーメントの計算式(Z
方向を表面垂直方向とする)は、
μ = ∫zρ(r)dr
を計算することによって求められます。積分はスラブモデルの場合、スラブ
の中心から、真空層の中心(スーパーセル全体の半分に相当)までの体積積分
です。
筆者は、この積分計算を、まず、バンド計算から求めた、逆空間での電子の
電荷密度ρ(G)を、フーリエ変換(注:高速FFTではない)してρ(r)を求め、
それをもとに上式に右辺の空間に関して(3次元)の積分を行ない、双極子モー
メントを求めました。これは、3次元空間に関してのメッシュの数にもよりま
すが、大変時間を要する計算となります。実際、Alphaチップ(Alpha21264)
搭載のワークステーションを使用しても、2、3日以上かかってしまいました。
これは甚だ計算時間を消費(浪費)した計算だと言えます。
ところが、よく考えてみると、これはもっとずっと簡単に計算可能であるこ
とが分かりました。きっかけは、昔JRCATの森川先生から貰ったプログラ
ムコードを見ていて、筆者と同じように双極子モーメントの計算をするルーチ
ンがあり、その内容が筆者のものと全然異なることに気付いたことでした。
上記の、μを求める式で、ρ(r)はフーリエ逆変換(ρ(G)→ρ(r)を逆変換
とする)を使って、
ρ(r) = Σρ(G)eiGr
と表現できます。これを、先のμを求める式に代入すると、
μ = ∫z(Σρ(G)eiGr)dr
となり、更に、
μ = Σρ(G)∫z(eiGr)dr
となります。上の式では、(定)積分部分は最早、数値積分に頼らずに解析
的に解けてしまいます(もう忘れかけているが、部分積分を使う)。つまり、
逆格子Gに関しての和のループだけで計算可能になってしまう訳です。解析的
の解く上で、上記の積分は、更に、Gr(ベクトル積)の部分が、
Gxx + Gyy + Gzzとなり、
∫z(eiGr)dr = ∫z(ei(Gxx + Gyy
+ Gzz))dxdydz
= ∫(eiGxx)dx∫(eiGyy)dy
∫z(eiGzz)dz
となります。
正に、解析的な手法に勝る、数値計算なしと言えます。
実際の計算、検証等は、学会(日本物理学会第55回年次大会)後に行なう
予定です。
(H)本日のバグ(レポート8/29、2000)
[目次]
今回もごく卑近な間違いです。バンド計算で得られた電子の電荷密度(逆空
間)を読み込み、それを実空間へフーリエ変換(注:高速フーリエではない)
して実空間の電荷密度にして、これを積分して全電荷数(電子数)を求める計
算を行なう計算で、間違っていました。
扱った系は表面系(スーパーセル+スラブモデル[*])で、最初はスーパーセル全体の電荷数
(電子数)を計算し、この段階では総電子数は、その系の真の電子数と十分な
精度に一致していました。ところが、スーパーセルの半分、スラブの中心から、
真空層の中心までを積分範囲として、電荷密度の和をとると、セル全体の総電
子数の半分にならず若干誤差が出ました。
系(総電子81個からなる)全体の総電子数
total chrage = 81.0000000025603
スラブの中心から、真空層の中心(系の半分に相当)までの総電子数
TOTAL CHRGE(FIX) = 40.6109851973210
上記値を見ると、総電子数が、40.5になっていませんでした。最初、この程
度は誤差として許容できるのではと思ったのですが、系全体で積分すると正し
く、総電子数は81(誤差:0.0000000025603)になるのに、積分領域を半分
にすると、0.1109851973210もの誤差が生じるのは、やはりおかしいというこ
とで、原因を追求することにしました。
最初、系が完全に対称になっていないため、スラブの中心の右側、左側(上
側、下側とも言えるが、左右で表現することにする)で電荷密度分布が対称に
なっていないのではないかと考えましたが、スラブでの原子の配置は左右で完
全に対象になっており、この配置で電荷密度分布が非対称になっていることは
あり得ないと判断しました(もしそれで非対称になる場合は、元のバンド計算
そのものがおかしいことになる)。
筆者の計算では、スラブの底を座標ゼロとして、スラブ+真空層からなる系
(スーパーセル)でバンド計算を行なっていましたが、これを真空層+スラブ
+真空層とし、スーパーセルの中心が、スラブの中心と一致するようにしてバ
ンド計算を行ない、それで得られた電荷密度で、上記積分計算を行なってみま
したが、結果は変わりませんでした。当然、バンド計算においても結果は、い
ずれの場合でも同じ結果(全エネルギー等)を与えます。
ここで、万策尽きかけたのですが、問題は積分の刻みの取り方にあるのでは
ないかと思い、今度は、積分の仕方に着目してみることにしました。
積分計算では、仮にZ方向を表面垂直方向とすると、フーリエ変換(注:高
速フーリエではない)した実空間電荷密度ρ(x,y,z)を、まずx,y方向で積分し、
その後、z方向で積分します(z方向の積分刻み数500)。実際の計算ループ
では、1〜500までの繰り返しを行ないますが、この場合、積分範囲をAと
すると、刻み幅を、hz=A/500とし、実際に積分を行なう座標は、z=(k-1)*hz、
としていました(kは繰り返しループでのループ変数)。
筆者は、z=k*hz、の場合の計算をしてみることにしました。そうすると積分
の結果は、
TOTAL CHRGE(FIX) = 40.3890148052776
となりました。これは、先の結果、
TOTAL CHRGE(FIX) = 40.6109851973210
と比べると、丁度両方を足すと、ほぼ正確な総電子数81になることが判明
しました。
系全体を積分した場合は、正しい総電子数が得られる。積分が1〜500と
0〜499で値が異なり、両方を足すとほぼ正確な総電子数になる。この段階
で、筆者は積分手法に重大な誤りがあることに、気付いてしまいました。これ
までの積分は、単に刻み幅分の長方形を足しているだけでした。つまり、積分
を、
TOTC = TOTC + CHG(I,J,K)
としていました。これは数値積分における、台形公式になっていません。台
形公式として積分するには、
TOTC = TOTC + (CHG(I,J,K) +CHG(I+1,J,K) )*0.5D0
としないといけません。このため積分計算の精度が悪いものになっていまし
た。この場合、系全体での積分では、電荷密度分布がスラブの中心から左右で
完全に対称になっているため、ちょうど右半分の区間の積分と左半分の区間の
積分の和が、上記の台形公式と等価となっているため正確な値が得られていま
した。
(予告)
(9/12、2000)この計算は、非常に効率の悪いものであることが判
明しました。わざわざ普通のフーリエ変換を行なって実空間での積分をする必
要がありませんでした。近々、これに関してのレポートをする予定です。現在、
筆者は大変多忙のため、暫くお待ち下さい。
(G)本日のバグ(レポート5/25、2000)
[目次]
また、久々にやらかしてしまいました(この”久々”にも[意味]あり)。今回の誤りはOpenMPによるSMP下での並
列計算で起こりました。
この前のOpenMP絡みの[バグ]以来、並列計算は行なっ
ていなかったのですが、計算速度向上のため、しばらくぶりに計算を行なって
みました。そうしたところ、計算結果がオリジナルの結果(OpenMPなしの単一
CPU動作による結果)と一致しないことが判明しました。
まず、OpenMPによる並列動作結果を以下に示します。
SUB-NAME = KBMSD ----------------- TIME = 0.000000
OCCUP2 : WIDTH= 5.000000000000000E-003
---------- THE FERMI ENERGY =-0.3609612356673470D-01 81.000000
SUB-NAME = FERMI ----------------- TIME = 0.000000
CALL FORCE
SUB-NAME = FORCE ----------------- TIME = 0.000000
SUB-NAME = CHAVER ----------------- TIME = 0.000000
SUB-NAME = XCFFT ----------------- TIME = 0.000000
SUB-NAME = FORLOC ----------------- TIME = 0.000000
MINUS CHG ICCCC = 0
TOTAL ENERGY FOR 1-TH ITERATION= -95.1142476 -0.9511425D+02
SUB-NAME = ENERGY ----------------- TIME = 0.000000
ITER= 1 ET(H)= 0.1000000D+06 ET(M)=********** DC= 0.5504872D-01
SUB-NAME = CONV2 ----------------- TIME = 0.000000
READ 2 --- REINIT OR NOT
READ 2 --- REINIT OR NOT 0
EVOUT ITER = 1
MINUS CHG ICCCC = 7
DTIME = 2.00000000000000
SUB-NAME = C3FFT ----------------- TIME = 0.000000
SUB-NAME = MSD ----------------- TIME = 0.000000
---------- THE FERMI ENERGY =-0.8970905953935529D-01 81.000000
SUB-NAME = FERMI ----------------- TIME = 0.000000
CALL FORZFB
SUB-NAME = FORCE ----------------- TIME = 0.000000
SUB-NAME = CHAVER ----------------- TIME = 0.000000
SUB-NAME = XCFFT ----------------- TIME = 0.000000
SUB-NAME = FORLOC ----------------- TIME = 0.000000
MINUS CHG ICCCC = 13
TOTAL ENERGY FOR 2-TH ITERATION=-103.3155412 -0.1033155D+03
SUB-NAME = ENERGY ----------------- TIME = 0.000000
ITER= 2 ET(H)= 0.8201294D+01 ET(M)=********** DC= 0.5515755D-01
SUB-NAME = CONV2 ----------------- TIME = 0.000000
MINUS CHG ICCCC = 7
---------- THE FERMI ENERGY =-0.4685106501394076D-01 81.000000
CALL FORZFB
MINUS CHG ICCCC = 23
TOTAL ENERGY FOR 3-TH ITERATION= -99.3989027 -0.9939890D+02
>> ETOOLD.LT.ETONEW <<
ETONEW-ETTOOLD= 3.91663855637013
ITER= 3 ET(H)= 0.1000000D+01 ET(M)=********** DC= 0.5920008D-01
MINUS CHG ICCCC = 7
---------- THE FERMI ENERGY =-0.2932751999252538D-01 81.000000
CALL FORZFB
次に、正しい(オリジナル)結果を示します。
SUB-NAME = KBMSD ----------------- TIME = 0.000000
OCCUP2 : WIDTH= 5.000000000000000E-003
---------- THE FERMI ENERGY =-0.3609612356673101D-01 81.000000
SUB-NAME = FERMI ----------------- TIME = 0.000000
CALL FORCE
SUB-NAME = FORCE ----------------- TIME = 0.000000
SUB-NAME = CHAVER ----------------- TIME = 0.000000
SUB-NAME = XCFFT ----------------- TIME = 0.000000
SUB-NAME = FORLOC ----------------- TIME = 0.000000
MINUS CHG ICCCC = 0
TOTAL ENERGY FOR 1-TH ITERATION= -83.4446440 -0.8344464D+02
SUB-NAME = ENERGY ----------------- TIME = 0.000000
ITER= 1 ET(H)= 0.1000000D+06 ET(M)=********** DC= 0.5504872D-01
SUB-NAME = CONV2 ----------------- TIME = 0.000000
READ 2 --- REINIT OR NOT
READ 2 --- REINIT OR NOT 0
EVOUT ITER = 1
MINUS CHG ICCCC = 7
DTIME = 2.00000000000000
SUB-NAME = C3FFT ----------------- TIME = 0.000000
SUB-NAME = MSD ----------------- TIME = 0.000000
---------- THE FERMI ENERGY =-0.8970905953933028D-01 81.000000
SUB-NAME = FERMI ----------------- TIME = 0.000000
CALL FORZFB
SUB-NAME = FORCE ----------------- TIME = 0.000000
SUB-NAME = CHAVER ----------------- TIME = 0.000000
SUB-NAME = XCFFT ----------------- TIME = 0.000000
SUB-NAME = FORLOC ----------------- TIME = 0.000000
MINUS CHG ICCCC = 13
TOTAL ENERGY FOR 2-TH ITERATION= -91.6459376 -0.9164594D+02
SUB-NAME = ENERGY ----------------- TIME = 0.000000
ITER= 2 ET(H)= 0.8201294D+01 ET(M)=********** DC= 0.5515755D-01
SUB-NAME = CONV2 ----------------- TIME = 0.000000
MINUS CHG ICCCC = 7
---------- THE FERMI ENERGY =-0.4685106501392522D-01 81.000000
CALL FORZFB
MINUS CHG ICCCC = 23
TOTAL ENERGY FOR 3-TH ITERATION= -87.7292991 -0.8772930D+02
>> ETOOLD.LT.ETONEW <<
ETONEW-ETTOOLD= 3.91663855636882
ITER= 3 ET(H)= 0.1000000D+01 ET(M)=********** DC= 0.5920008D-01
MINUS CHG ICCCC = 7
---------- THE FERMI ENERGY =-0.2932751999250589D-01 81.000000
CALL FORZFB
(説明)
深紅色と深桃色
で、それぞれ示されているのが、全エネルギーの値です。お分かりの
ように、全エネルギーの値は、それぞれ全く一致していません。
以前の[バグ]が解決した時点では、オリジナルの結果
との比較を怠っていました。並列計算で、1CPUのみの実行結果と、多CP
U(2ないし4)での実行結果との比較(これらの結果は一致)したことで、”
よし”としていました。この点で詰めが甘かったと言えます。
不一致の原因究明の作業過程で浮かび上がった事実は、
- OpenMPによる計算では、1CPUのみの計算結果と、多CPUの計算結
果は一致。
- OpenMPによる計算結果と、オリジナルの結果は全エネルギーが不一致。
- 全エネルギーの結果は第一回目のイタレーションの結果から不一致。
- オリジナル計算のソースプログラムを、OpenMP並列計算を行なっている
ディレクトリに複写し、OpenMP計算と全く同じ環境(違いは、並列動作しない
だけ)で(単一CPUで)計算しても、計算結果は一致しない(当然、この結
果はオリジナルの結果とは一致した)。
- OpenMP用のソースプログラムを、OpenMP用のコンパイルオプション(-
omp)なしで、コンパイルしても結果は変わらなかった。
- OpenMPによる並列計算結果と、オリジナルの結果を、よくよく比べてみ
ると、全エネルギーは一致していないが、フェルミエネルギーの値(深橙色で表示)は、互いに一致していることが分
かった(重要)。
特に、最後の項目は非常に重要です。初期の段階から、第一回目のイタレー
ションでの全エネルギーが一致しないことは判明していましたが、第一回目の
フェルミエネルギーの値は一致していました(非常に小さな値の部分での不一
致は除く)。筆者は、この段階で、最初のフェルミエネルギーの計算をするサ
ブルーチンまでは正しく計算が行なわれていると判断しましたが、それ以降に
ついては、全エネルギーが一致しないので、フェルミエネルギーも、これ以降
は一致していないだろうと思い込んでしまいました。この計算では、全エネル
ギーの計算の前にフェルミエネルギーの計算が行なわれます(そうでないと、
全エネルギーは計算できない)。
この判断は完全な誤りで、よくよく並列計算の結果と、オリジナルの結果を
比べてみると、その後のイタレーションでもフェルミエネルギーの値は誤差を
除いて完全に一致していました。これは、非常に重要なことで、普通数イタレー
ション計算を経た後で、全エネルギーが一致しないのに、フェルミエネルギー
だけが一致することはありえません。そこで、各イタレーションでの全エネル
ギーの値とフェルミエネルギーの値を以下に示したいと思います。
まず、OpenMPによる並列計算の場合を示します。
TOTAL ENERGY FOR 1-TH ITERATION= -95.1142476 -0.9511425D+02
TOTAL ENERGY FOR 2-TH ITERATION=-103.3155412 -0.1033155D+03
TOTAL ENERGY FOR 3-TH ITERATION= -99.3989027 -0.9939890D+02
TOTAL ENERGY FOR 4-TH ITERATION= -97.7885536 -0.9778855D+02
TOTAL ENERGY FOR 5-TH ITERATION= -97.6782727 -0.9767827D+02
1-TH ----- THE FERMI ENERGY =-0.3609612356673470D-01 81.000000
2-TH ----- THE FERMI ENERGY =-0.8970905953935529D-01 81.000000
3-TH ----- THE FERMI ENERGY =-0.4685106501394076D-01 81.000000
4-TH ----- THE FERMI ENERGY =-0.2932751999252538D-01 81.000000
5-TH ----- THE FERMI ENERGY =-0.1644041912340696D-01 81.000000
6-TH ----- THE FERMI ENERGY =-0.6217050465534285D-02 81.000000
次に、オリジナルの場合を示します。
TOTAL ENERGY FOR 1-TH ITERATION= -83.4446440 -0.8344464D+02
TOTAL ENERGY FOR 2-TH ITERATION= -91.6459376 -0.9164594D+02
TOTAL ENERGY FOR 3-TH ITERATION= -87.7292991 -0.8772930D+02
TOTAL ENERGY FOR 4-TH ITERATION= -86.1189500 -0.8611895D+02
TOTAL ENERGY FOR 5-TH ITERATION= -86.0086691 -0.8600867D+02
1-TH ----- THE FERMI ENERGY =-0.3609612356673101D-01 81.000000
2-TH ----- THE FERMI ENERGY =-0.8970905953933028D-01 81.000000
3-TH ----- THE FERMI ENERGY =-0.4685106501392522D-01 81.000000
4-TH ----- THE FERMI ENERGY =-0.2932751999250589D-01 81.000000
5-TH ----- THE FERMI ENERGY =-0.1644041912338591D-01 81.000000
6-TH ----- THE FERMI ENERGY =-0.6217050465513631D-02 81.000000
以上を比較してみると、確かに、小数点以下の非常に小さな部分を除けば、
フェルミエネルギーの値は一致し、全エネルギーの値は異なっています。前述
のように全エネルギーが異なる場合、バンド構造そのものも異なるはずで、フェ
ルミエネルギーは一致しないはずなのに、実際は一致しています。この場合考
えられるのが、バンド計算そのもの(セルフコンシステントな計算過程:つま
りイタレーション)には影響しない部分で不一致が起こっていることが考えら
れます。そこで、OpenMPによる並列計算とオリジナルな計算での各イタレーショ
ンでの全エネルギーの値の差をとってみると、それはイタレーション回数に関
係なく、11.6696036(絶対値)という一定値であることが分かりました。
(原因究明)
この、11.6696036という値は、調べてみると、部分内殻補正で使用していた
補正項の値であることが判明しました。この補正項は、バンド計算の最初で設
定されるもので、値そのものは全エネルギーの計算の時に、全エネルギーの値
に足し込まれるだけで、バンド計算そのものの結果には全く影響を与えないも
のです。前回の[バグ]の時にデバッグ中に、この計算部
分がバグの原因ではと削除しておいたのを完全に忘れていました。元に戻さず
に計算を行なったため、全エネルギーの値が一致しないように見え、並列計算
に深刻なバグがあるのではと慌てた次第です。
実際は、バグとしてはそう深刻なものではなく、ほんの数行の修正(今回の
場合は削除した部分の追加)で、問題を解決することができました。問題は、
むしろバグを直したと思って、2、3ヶ月もほったらかしておくと、何をした
かを忘れてしまうことです。作業記録をきちんと残し、やり残したことや、無
用な変更、削除を元に戻しておくことを怠らないよう(うっかり忘れないよう)
にし、その時できることは、その時に完結させておく(記録も残す)ようにし
ましょう。
(F)本日のバグ(レポート2/24、2000)
[目次]
今回のバグは、部分内殻補正絡みもの
です。
筆者のプログラムで、部分内殻補正の計算を行なう場合、扱う物質や系によっ
てストレスの値が発散する(異常に大きな数になる)ことがありました。ただ、
扱っていた系が表面系だったり(普通、表面系ではユニットセルにかかる圧力
〔ストレス〕は使わない。表面ストレスを求めようという場合は別だが)、そ
の時点でストレスは必要なかったりで、さして気に止めていませんでした。
しかし、やはりしっくりこないこともあり、原因を突き止めることにしまし
た。経験上バルク系では、ストレスが異常に起こることはほとんどなかったの
ですが、最近テストとして計算していたバルク系で、ストレス値が10
18もの値になる事態に遭遇していたので、これをデバッグしてみること
にしました。以下に、異常なストレス値が出る場合の出力結果を示します。
TOTAL STR 1 1 = -0.106726751049972
2 = 1.114843552513939E-003
3 = 3.786085519029700E-004
4 = -0.107180229756837
5 = -6.187637920119319E-006
6 = -0.107625292219891
REAL TOTAL CHARGE = 8.00229133076466 IN XSTPC
ICOUNT = 2244 XSTPC END
STMQ,SS1,SSX,ZXXX(1) = 4.499630388065659E-003 0.000000000000000E+000
7.977901970862270E+020 (-1.118441627010112E+022,379875.028700487)
ZCHG(1),ZRHPC(1),ZVXC(1),ZRRPC(1),ZEXC(1) =
(6.713436944043497E-002,0.000000000000000E+000)
(4.196147755972463E-003,0.000000000000000E+000)
(-0.279550593402226,0.000000000000000E+000)
(0.000000000000000E+000,0.000000000000000E+000)
(-0.216469172365056,0.000000000000000E+000)
TOTAL STR 2 1 = 7.977901970862270E+020
2 = 0.000000000000000E+000
3 = 0.000000000000000E+000
4 = 7.977901970862274E+020
5 = 0.000000000000000E+000
6 = 7.977901970862264E+020
ZVX,ZEX = -8.052755104079717E+020 -3.244955581708985E-002
STM1(XY)= 1.128607285201954E-002 -0.163484318525950
STU,V,W = 0.126066049507907 -5.284113835626290E-004
-8.052755104079717E+020
STOT = -8.052755104079717E+020
TOTAL STR 3 1 = -8.052755104079578E+020
2 = 5.204170427930421E-018
3 = 2.981555974335137E-018
4 = -8.052755104079581E+020
5 = 5.175371307723775E-019
6 = -8.052755104079573E+020
SIGNL 1 1 = 0.221485143478849
2 = 0.000000000000000E+000
3 = 0.000000000000000E+000
4 = 0.221485143478849
5 = 0.000000000000000E+000
6 = 0.221485143478849
SIGNL 2 1 = -3.008891331718425E-002
2 = -4.277013915213738E-005
3 = -1.448585533509181E-005
4 = -3.008246681184892E-002
5 = 6.375125928747571E-007
6 = -3.007457410262290E-002
SIGNL 3 1 = -6.993430363272542E-002
2 = 9.378111455720368E-004
3 = 3.224221320190277E-004
4 = -7.021242131084532E-002
5 = 3.474492615092482E-006
6 = -7.048630456989730E-002
SIGNL 4 1 = -0.135626705862546
2 = 9.203622245516506E-004
3 = 3.093997338064650E-004
4 = -0.136006849184124
5 = -1.467699662472126E-005
6 = -0.136396710849138
SIGNL 5 1 = 0.135041992087479
2 = -2.647062024157973E-003
3 = -9.026027297115934E-004
4 = 0.136016126412455
5 = 9.309930437597757E-006
6 = 0.136972147395874
SIGEWA 1 = 5.254142857619389E-002
SIGEWA 2 = 7.470500746495105E-018
SIGEWA 3 = -4.548484389417145E-018
SIGEWA 4 = 5.254142857619389E-002
SIGEWA 5 = 1.892189202705232E-018
SIGEWA 6 = 5.254142857619388E-002
SIGNL 1 = 0.120877212753873
SIGNL 2 = -8.316587931864228E-004
SIGNL 3 = -2.852667192211925E-004
SIGNL 4 = 0.121199532584486
SIGNL 5 = -1.255060979156260E-006
SIGNL 6 = 0.121499701353065
TOTAL STR 4 1 = 0.000000000000000E+000
2 = -8.316587931864153E-004
3 = -2.852667192211971E-004
4 = 0.000000000000000E+000
5 = -1.255060979154368E-006
6 = 0.000000000000000E+000
ETOT1,TOTCH = 0.316363262756181 8.00000000000000
TOTAL STR 5 1 = 0.000000000000000E+000
2 = 0.000000000000000E+000
3 = 0.000000000000000E+000
4 = 0.000000000000000E+000
5 = 0.000000000000000E+000
6 = 0.000000000000000E+000
TOTAL SUMM 1 = -7.485313321730875E+018
2 = 2.831847593275287E-004
3 = 9.334183268177589E-005
4 = -7.485313321730745E+018
5 = -7.442698899273169E-006
6 = -7.485313321731027E+018
TOTAL SUMM OP1 = -7.485313321730884E+018
2 = 7.058607893785836E-023
3 = 0.000000000000000E+000
4 = -7.485313321730884E+018
5 = -2.258754526011467E-021
6 = -7.485313321730885E+018
CALL FORLOC AND MD
NOW MD SET EPP 11
EPP(1,1) = 1.00000000000000 0.000000000000000E+000
EPP(1,2) = 0.000000000000000E+000 0.000000000000000E+000
EPP(1,3) = 0.000000000000000E+000 0.000000000000000E+000
EPP(2,1) = 0.000000000000000E+000 0.000000000000000E+000
EPP(2,2) = 1.00000000000000 0.000000000000000E+000
EPP(2,3) = 0.000000000000000E+000 0.000000000000000E+000
EPP(3,1) = 0.000000000000000E+000 0.000000000000000E+000
EPP(3,2) = 0.000000000000000E+000 0.000000000000000E+000
EPP(3,3) = 1.00000000000000 0.000000000000000E+000
TOTAL ENERGY FOR 11-TH ITERATION= -32.4511839 -0.3245118D+02
MIXING RATE = 0.800000000000000
ITER= 11 ET(H)= 0.1239419D-01 ET(M)=337.122064 DC= 0.1087433D-01
もうデバッグ用の計算になっているので、11回イタレーションを進めた段
階でプログラムを止めています(計算は収束していない)。深紅色(CRIMSON)
で表示されている部分が異常に大きな値をしめしているストレス値です。もと
もとデバッグも考慮して、各計算途中段階でのストレスの値を出力するように
していました(上記ではデバッグ用に新たに出力させたものもある)。この結
果から、かなり早い段階(”TOTAL STR 2 1 = ”と
表示されたところ)で、値が異常になっていることが分かります。
筆者がまず最初に注目したのは、この”TOTAL STR 2
1 = ”の段階の表示部分で、これらの変数(配列変数)は最初の値(つま
りA(1)とかB(1))しか表示していないことでした。筆者のバンド計算プログラ
ムでは、逆格子ベクトルは大きさ順に並べるので、最初の配列変数の値(例
ZCHG(1))は、G=0での値となります。G=0では発散項などが生じる場合
があり、扱いに特別な考慮が必要な場合があり、ストレスの計算でも変数の1
番目は別途計算する場合があり、それがTOTAL STR 2
1の部分だった訳です。
この段階で値がおかしいので、さらに詳しく値を表示させることにし、
STMQ,SS1,SSX,ZXXX(1) = 4.499630388065659E-003 0.000000000000000E+000
7.977901970862270E+020 (-1.118441627010112E+022,379875.028700487)
ZCHG(1),ZRHPC(1),ZVXC(1),ZRRPC(1),ZEXC(1) =
(6.713436944043497E-002,0.000000000000000E+000)
(4.196147755972463E-003,0.000000000000000E+000)
(-0.279550593402226,0.000000000000000E+000)
(0.000000000000000E+000,0.000000000000000E+000)
(-0.216469172365056,0.000000000000000E+000)
という結果を得ました。この計算結果から逆格子空間での電荷密度、部分内
殻電荷密度、交換相関項の値(ZCHG(1),ZRHPC(1),ZVXC(1),ZRRPC(1),ZEXC(1))
には異常はなく、SSX、そしてZXXX(1)の値に問題があることが分かります。
特に問題となるのが、ZXXX(1)で、これと連動してSSXという値もおかしくな
ります。ストレス値の異常の根本原因は、変数ZXXXにありそうだということで、
今度はZXXX(1)だけでなく、ZXXXの全ての値を表示させてみました。結果とし
てZXXXは全ての値が異常(発散している)であることが分かりました。
ZXXXは、部分内殻補正導入によって生じる、新たなストレス項(交換相関項
が関わる)を計算するための変数で、それは以下の実空間での交換相関項に関
しての計算、
C -- X ------------
DO 1020 I=1,KSUM
RS=CO2*( 1.0D0/CHGB1(I) )**THIRD
RH1 = -0.458D0*CO3/RS-(0.44D0*CO3*RS + 3.432D0)/
& (RS+7.8D0)**2
RH2 = -0.458D0/RS-0.44D0/(RS+7.8D0)
C
RHV(I)=RH1 - RH2
ZV1D(I)=ZRX(I)*(RHV(I))/CHGB1(I)
1020 CONTINUE
の、変数ZV1Dをフーリエ変換(実空間→逆空間をフーリエ変換と定義)した
ものがZXXXとなります。問題なのは、深紅色で示した、1/CHGB1(I)の部分で、CHGB1(I)は実空間での電荷密
度です。実空間の電荷密度は、逆格子空間で計算された電荷密度ZCHGを逆フー
リエ変換して求めます。部分内殻補正を考慮した場合は、ZCHG+ZRHPCと部分内
殻補正電荷密度がZCHGに加わります。普通、実空間電荷密度が負の値になるこ
とはありえないのですが、場合によっては負の値になってしまうことがありま
す。それは系が表面系で、真空領域がある場合(どうしてもフーリエ変換等の
過程で誤差が入り込む)や、今回の部分内殻補正を考慮した場合です。部分内
殻補正用の電荷密度を加えて、(逆)フーリエ変換する間に、何らかの誤差が
入り込んで電荷密度が負になる場合があります(どういう兼ね合いでそうなる
かは更に原因調査中)。
そして実空間での電荷密度(部分内殻補正電荷密度込み)CHGB1が負の値に
なった場合、CHGB1の値を10-40にする処理をしていました。この
ため今回の場合、1/CHGB1が発散してしまう事態となった訳です。分母の項は、
この発散を打ち消す程の値にならないことも判明しました。分母の項は
RS=CO2*( 1.0D0/CHGB1(I) )**THIRDの逆数
のオーダーなのですが、このTHIRDが1/
3のため、分母はCHGB1の1/3乗のオーダーで、1/CHGB1をかけることにより発散してしまいます。
対処の仕方は、RCHGB(I) = 1.0D0/CHGB1(I)という変数を定義し、CHGB1が負
の値になった場合、RCHGBはゼロになるようにしておきます。そして、
C ZV1D(I)=ZRX(I)*(RHV(I))/CHGB1(I) <-- 2/18, 2000
ZV1D(I)=ZRX(I)*(RHV(I))*RCHGB(I)
とすれば、ZXXX(ZV1Dをフーリエ変換)の発散を防ぐことができます。
(E)本日のバグ(レポート1/17、2000)
[目次]
今回のバグは比較的軽微なものです(場合によっては深刻)。
問題はサブルーチンPCC
内の配列変数CHGPCです。この変数は、サブルーチンPCC内で定義されてい
る局所的な配列変数で、READ文により読み込まれた部分内殻補正用電荷密度の
総電荷数が格納されます。問題は、この読み込みは最初のイタレーションでし
か行なわれないのですが、CHGPCはその後のイタレーションでも使用されます。
普通、その場合は、サブルーチンの引数としてCHGPCは
メインプログラム側へ引き継がれるようになっていなければならないのに、
実際は、SUBROUTINE PCC(KTPCC,RHPC,SCHGPC,EPC,PCM)と、CHGPCは()内の引
数の中に存在しません。
このため、最初のイタレーション以降、CHGPC内の値は不定となってしまい、
正しい値が保持される保証は全くありませんでした。実際に調べてみると
CHGPCの値は、DEC(現COMPAQ)のAlphaマシン、富士通のVPP(並
列計算)では最初のイタレーションの値が正しく保持されていました。但しこ
れは非常に好運だったと言えます。また、CHGPC自身も他のサブルーチンで使
用されておらず(されていたら引数を受け渡していたはず)、たとえ間違った
値になったとしても深刻な問題を起こす変数ではありませんでした。逆にこの
ため、この問題点に気付くのが遅れたとも言えます。
問題発見の端諸は、旧DECのSMPマシン上でOpenMPプログラムのテスト
中に、並列計算が途中で止まってしまい、止まる箇所がサブルーチンPC
Cであったことです。詳しく問題原因を調査した結果、上記のバグが判明
しました。並列計算が止まる原因もCHGPCの値が、OpenMPによる並列計算では
保持されず(VPPでの並列計算では保持【単なる偶然?】、但し全ての場合
はチェックしていない)、これが元でプログラムが異常終了してしまったよう
です。CHGPCをメインルーチンに引き渡すようにすると並列計算は問題なく行
なえるようになりました。
今回のバグは結果として軽微(OpenMPによる並列計算では障害があったが)
でしたが、まかり間違えば大きなバグに繋がる可能性があります。局所変数、
大域変数の定義、動作等のチェックはちゃんとするようにしましょう。コンパ
イラによってはオプション等で引数等のチェックは可能ですが、サブルーチン
内で定義された(配列)変数が局所的か大域的かまでは普通判断できません
(人が見るしかない)。
(4/27、2009追記)今年度、スーパーコンピュータ更新に伴い、本格
的なOpenMP化作業を始めた。情けないことに再び、CHGPCに関する同じ間違い
を犯してしまう。CHGPCに関して上記と同じ修正を行なうことで問題は解決す
る。9年経っても進歩なし。そして過去の教訓が全く生かされていな
い(←強く反省すべき点)。
(12/1、2011追追記)再び同じ間違いをやらかす。悲しい。上記(4/
27、2009)に関する修正が行なわれていいないプログラム(コード)が
まだ残っており、実行中にエラーで計算が止まってしまった。早速、修正を施
すが、全くもってなってない話(←改めて強く反省)。
(5/22、2012追追追記)まだ間違えたままのプログラムが残っていた。
(2/22、2016追追追追記)いつまで追記は続くのだろう。ただ今回、
問題が顕在化するような使用をほとんどしたことがない版のプログラムだった
ので、仕方のない部分もある。これまで磁性関連のプログラムで、構造最適化
等まで含めた計算をほとんど(と言うか全く)行なっていなかったので、問題
が顕在化(気付く)するのが本日になってしまった(構造最適化させない限り
問題なかった。→構造最適化させた途端異常終了して気付いた次第)。
(9/28、2022追追追追追記)また間違いから数値の発散が起こる(磁性の計算)。PCC(部分内殻補正)なしの計算で電荷の値が発散表示された。プログラム冒頭で、SCHGPC = 0.0D0とすると解決。6年経っても相変わらずな状況に愕然とする。
(D)本日のバグ(レポート4/20、1999)
[目次]
3カ月ぶりのバグレポートですが、今回は、これまでのバグの中でも、最大
級に深刻なものです。
それは以下のようなものです。
サブルーチンFORLOC
(既に修正済み)において、これまでは、
C FORCE2 AND PRESSURE ADDITION SSN(KNG1)
DO 161 IA=1,KATM
DO 162 I=1,KG
FX = GX(I)
FY = GY(I)
FZ = GZ(I)
ZFORT =CDEXP(-ZI*(CATX(IA)*FX+CATY(IA)*FY+CATZ(IA)*FZ))
ZFTMP =ZFORT *PSC(I,KFTYPE(IA))*DCONJG(ZCHG(I))
ZFORC2(IA,1)=ZFORC2(IA,1)+GX(I)*ZFTMP
ZFORC2(IA,2)=ZFORC2(IA,2)+GY(I)*ZFTMP
ZFORC2(IA,3)=ZFORC2(IA,3)+GZ(I)*ZFTMP
162 CONTINUE
IF (KTPCC(KFTYPE(IA)).EQ.1) THEN
DO 1000 N=1,6
ZFPC(N)=DCMPLX(0.0D0,0.0D0)
1000 CONTINUE
DO 200 I=1,KG
ZFORT =CDEXP(-ZI*(CATX(IA)*FX+CATY(IA)*FY+CATZ(IA)*FZ))
ZFTMP =ZFORT *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXC(I))
ZFPC(1)=ZFPC(1)+GX(I)*ZFTMP
ZFPC(2)=ZFPC(2)+GY(I)*ZFTMP
ZFPC(3)=ZFPC(3)+GZ(I)*ZFTMP
ZFTMP =ZFORT *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXCPC(I))
ZFPC(4)=ZFPC(4)+GX(I)*ZFTMP
ZFPC(5)=ZFPC(5)+GY(I)*ZFTMP
ZFPC(6)=ZFPC(6)+GZ(I)*ZFTMP
200 CONTINUE
としていました。問題は、DO 162ループ内のFX、FY、FZです。
これはフォームファクターを計算するための変数なのですが、これがDO 2
00ループ内で定義されていません。DO 161とDO 200は完全に独立
したループで、DO 200ループではFX、FY、FZを再定義しなければ
ならないのに、それを怠っていました。これでは正しい計算ができるはずがあ
りません。
修正は、
C FORCE2 AND PRESSURE ADDITION SSN(KNG1)
DO 161 IA=1,KATM
DO 162 I=1,KG
FX = GX(I)
FY = GY(I)
FZ = GZ(I)
ZFORT =CDEXP(-ZI*(CATX(IA)*FX+CATY(IA)*FY+CATZ(IA)*FZ))
ZFTMP =ZFORT *PSC(I,KFTYPE(IA))*DCONJG(ZCHG(I))
ZFORC2(IA,1)=ZFORC2(IA,1)+GX(I)*ZFTMP
ZFORC2(IA,2)=ZFORC2(IA,2)+GY(I)*ZFTMP
ZFORC2(IA,3)=ZFORC2(IA,3)+GZ(I)*ZFTMP
162 CONTINUE
IF (KTPCC(KFTYPE(IA)).EQ.1) THEN
DO 1000 N=1,6
ZFPC(N)=DCMPLX(0.0D0,0.0D0)
1000 CONTINUE
DO 200 I=1,KG
FX = GX(I)
FY = GY(I)
FZ = GZ(I)
ZFORT =CDEXP(-ZI*(CATX(IA)*FX+CATY(IA)*FY+CATZ(IA)*FZ))
ZFTMP =ZFORT *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXC(I))
ZFPC(1)=ZFPC(1)+GX(I)*ZFTMP
ZFPC(2)=ZFPC(2)+GY(I)*ZFTMP
ZFPC(3)=ZFPC(3)+GZ(I)*ZFTMP
ZFTMP =ZFORT *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXCPC(I))
ZFPC(4)=ZFPC(4)+GX(I)*ZFTMP
ZFPC(5)=ZFPC(5)+GY(I)*ZFTMP
ZFPC(6)=ZFPC(6)+GZ(I)*ZFTMP
200 CONTINUE
です。DO 200ループ内でFX、FY、FZを再定義します。このルー
プはPCC(部分内殻補正)に関しての
力の計算を行なっている部分です。
もし、ZFORTの計算で、
ZFORT =CDEXP(-ZI*(CATX(IA)*GX(I)+CATY(IA)*GY(I)+CATZ(IA)*GZ(I)))
としていたら、この間違いはなかったのですが、わざわざFX、FY、FZ
に代入してZFORTを計算するのには、確か意味がありました(多分、ベクトル
計算の上でこのようにした方が、計算上有利だったように憶えているのですが
確かではありません。現在調査中)。
調べてみると、古いバージョンのプログラムでは変数ZFORTが、配列変数ZFORT(I)になっていたことが分かりました。以下にそれを示します。
C FORCE2 AND PRESSURE ADDITION SSN(KNG1) Old version
DO 161 IA=1,KATM
DO 162 I=1,KG
FX = GX(I)
FY = GY(I)
FZ = GZ(I)
ZFORT(I) =CDEXP(-ZI*(CATX(IA)*FX+CATY(IA)*FY+CATZ(IA)*FZ))
ZFTMP =ZFORT(I) *PSC(I,KFTYPE(IA))*DCONJG(ZCHG(I))
C*****ZFTMP =ZFORM(I,IA)*PSC(I,KFTYPE(IA))*DCONJG(ZCHG(I))
ZFORC2(IA,1)=ZFORC2(IA,1)+GX(I)*ZFTMP
ZFORC2(IA,2)=ZFORC2(IA,2)+GY(I)*ZFTMP
ZFORC2(IA,3)=ZFORC2(IA,3)+GZ(I)*ZFTMP
162 CONTINUE
IF (KTPCC(KFTYPE(IA)).EQ.1) THEN
DO 1000 N=1,6
ZFPC(N)=DCMPLX(0.0D0,0.0D0)
1000 CONTINUE
DO 200 I=1,KG
ZFTMP =ZFORT(I) *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXC(I))
C***** ZFTMP =ZFORM(I,IA)*RHPCG(I,KFTYPE(IA))*DCONJG(ZVXC(I))
ZFPC(1)=ZFPC(1)+GX(I)*ZFTMP
ZFPC(2)=ZFPC(2)+GY(I)*ZFTMP
ZFPC(3)=ZFPC(3)+GZ(I)*ZFTMP
ZFTMP =ZFORT(I) *RHPCG(I,KFTYPE(IA))*DCONJG(ZVXCPC(I))
C***** ZFTMP =ZFORM(I,IA)*RHPCG(I,KFTYPE(IA))*DCONJG(ZVXCPC(I))
ZFPC(4)=ZFPC(4)+GX(I)*ZFTMP
ZFPC(5)=ZFPC(5)+GY(I)*ZFTMP
ZFPC(6)=ZFPC(6)+GZ(I)*ZFTMP
200 CONTINUE
DO 162もDO 200ループも変数IA(ユニットセル内の原子数に関し
て)のDO 161ループの内側にあるので、ZFORT(I)(で十分)となります。
注釈でのZFORT(I,IA)から、更に古いプログラムでは変数I(逆格子数)及びIA
(原子数)の配列であることが分かります。これから、ZFORT(I)からZFORTへ
の変更は、ベクトル化云々より、メモリを節約するためだったと考えられます。
そのための作業過程(ZFORT(I)→ZFORT)で、FX、FY、FZの再定義を忘
れたため、このようなバグが生じてしまいました。
以前(数年前)から、PCCに関しての力には、虚数の力が出てきたり、対
称性から考えてありえない力が出てきたりしていました。筆者は、それを負の
電荷密度(PCCを導入すると動径方向の実空間電荷をフーリエ変換し、再度
実空間電荷に逆フーリエする過程で出てくる)によるものだと解釈していまし
た。実際それによる影響もあるのですが、もっと重大で深刻な問題が潜んでい
ました。PCC版を導入してから数年以上経つ今の今まで全く気付きんません
でした。
このバグ発見の経緯は、GGA導入(ま
だ内容はできていません。→一応出来ていますがまだ未完成〔4/21、20
10〕)に向けて、どうも力の挙動がおかしいと思い、以前からPCCを考慮
していた計算でも力に賦に落ちないところがあったので、まずPCCのみ考慮
したバージョンで、簡単な系でテストをしていたら、バグに気付いた次第です。
テストは、Alを仮想的なBCC構造として、立方体のユニットセルを考え、
その中に二つのAl原子を置いて、111方向にそれを近付けたり、遠ざけた場
合(対称性からは近付けたことと同じ)での力の値を調べました。この場合、
Al原子は111方向上にあれば、どのような位置同士であっても、111方向
に同じ大きさの力(方向は互いに反対)を受けます。PCCを考慮しない場合
は、そうなるのですが、PCC版ではそうなりませんでした。
これは何か、GGA導入以前にPCCによる力の計算に深刻な問題があると
判断し、更にしろいろテストしていく内にバグを発見しました。バグそのもの
はさして複雑でも、見つけ難いものでもなく、非常に単純なものでした。しか
しながら、これによる影響は大変深刻です。少なくとも、これまで行なってき
たPCC考慮版の計算で、構造最適化を施したものの信頼性が(完全に駄目で
はないにしろ)大分落ちてしまいます。更に、現在保有するプログラムを全て、
修正しなければなりません。この作業は見落としなく、全てのプログラム(い
ろいろなマシンに分散して存在)を修正する作業は大変なものです。
(4/22、1999)現在、修正作業を行なっています。プログラムはい
ろいろなディレクトリに散らばって存在するので、作業はやはり大変です。
今回の教訓としては、以前のリポートでも書いているのですが、”おかしい”、”
何か釈然としない”、”計算結果がどうもしっくりいかない”と思ったら甘い
解釈はしないで、徹底的に調べてみるというものです。
(C)本日のバグ(レポート1/20、1999)
[目次]
今回はよくやる典型的な間違い(バグ)です。
以下のルーチンの部分に間違いがありました。
DO 2000 JBAN=1,KEG
DO 2010 IBAN=1,KEG
IF (IBAN.EQ.JBAN) THEN
DO 2100 J=1,IIBA
DO 2110 I=1,IIBA
I1=NBASE(I,NNN)
J1=NBASE(J,NNN)
CALL HPDF(HP,HD,HF,VP,VD,VF
& ,IA,NSEK,I,J)
Z(IBAN,JBAN)=Z(IBAN,JBAN) + EKO(IBAN,NNN)
& + DCONJG(ZZZ(I,IBAN,NNN))*RAA(I,NNN)*QC(I,J)*RAA(J,NNN)
& *HP*ZZZ(J,IBAN,NNN)*PC
& *ZFM2(I1,IA)*DCONJG(ZFM2(J1,IA))
2110 CONTINUE
2100 CONTINUE
2010 CONTINUE
2000 CONTINUE
上記ルーチンで、IBAN,JBANはバンド(インデックス)に関してのループ、
I,Jは平面波数に関してのループで、EKOはバンド計算におけるエネルギー固有
値です。そしてこのEKOを配列変数Zに足し込んでいるのですが、既に分った人
もいると思いますが、このEKOの位置に問題があります。このルーチンでは、
バンド数×バンド数の行列Zを作り、それを最終的に対角化(固有値問題、Zに
対する固有値〔先のバンド計算による固有値とは別物〕を求める)することを
目的としていました。対角項にはバンド計算によるエネルギー固有値も和の一
部として含んでいます。
ところが、実際対角化して得られた固有値は、とんでもなく大きな値になっ
てしまいました。原因を突きとめるため、上記ルーチンのHPの値をゼロにして
みましたが、状況は変わりませんでした。更に、以下のようにルーチンを変更
しても状況は依然変わりませんでした。
DO 2000 JBAN=1,KEG
DO 2010 IBAN=1,KEG
IF (IBAN.EQ.JBAN) THEN
DO 2100 J=1,IIBA
DO 2110 I=1,IIBA
I1=NBASE(I,NNN)
J1=NBASE(J,NNN)
C CALL HPDF(HP,HD,HF,VP,VD,VF
C & ,IA,NSEK,I,J)
Z(IBAN,JBAN)=Z(IBAN,JBAN) + EKO(IBAN,NNN)
2110 CONTINUE
2100 CONTINUE
2010 CONTINUE
2000 CONTINUE
この段階でどうしてだろうと頭を抱えそうになったのですが、他のループで
ZにEKOの値のみを代入して(上記ルーチンは動かなくする)対角化すると正し
い結果が出ました。そのルーチンは、
DO 272 I=1,KEG2
DO 273 J=1,KEG2
Z(J,I)=CMPLX(0.0,0.0)
IF (I.EQ.J) THEN
Z(J,I)=EKO(I,NNN)
END IF
273 CONTINUE
272 CONTINUE
としていました。ようやくこのルーチンと、先のルーチンを比較して筆者は
間違いに気付きました。問題は、
DO 2100 J=1,IIBA
DO 2110 I=1,IIBA
I1=NBASE(I,NNN)
J1=NBASE(J,NNN)
C CALL HPDF(HP,HD,HF,VP,VD,VF
C & ,IA,NSEK,I,J)
Z(IBAN,JBAN)=Z(IBAN,JBAN) + EKO(IBAN,NNN)
2110 CONTINUE
2100 CONTINUE
で、平面波数の2重ループ内において、エネルギー固有値EKOを足していま
した。つまりバンド×バンドに関しての行列の1つの対角要素に平面波数×平
面波数分のEKOの和が代入されていた訳です。これでは対角化による固有値が
とんでもなく大きな値(発散してもおかしくなかった)になるのも当然です。
この段階になるまで気付けなかったのがちょっと情けないです。
ただ、この手の和をとるとき、とるべき変数の位置を間違えるのは、よく犯
す間違いの典型例と言えます。間違い方としては、上記のように和をとる必要
のないループにその変数を置いてしまう場合と、和ととるべきループ以外の場
所にその変数を置いてしまう場合などです。前者では、大抵その変数は、その
和のループ内では定数和と同等となり、非常に大きな絶対値(大き過ぎれば発
散してエラーになる。今回は発散するまでには至らなかった)になって後々悪
さをします。
このように、和をとる時の変数位置の間違いは筆者も過去何度も経験してい
るのですが、またやってしまいました。変数の位置のチェックは怠らないよう
にしましょう。
(B)本日のバグ(レポート11/18、1998)
[目次]
今回も単純なミスです。
新しい系の計算をしようとして、擬ポテンシャル、初期設定用座標データな
どを用意して、テスト計算していたら間違っちゃいました。
最初、テスト計算で計算が4、5回イタレーションを回した段階で破綻(全
エネルギー発散)しました。この時点では、よくあることだと思い、初期設定
(波動関数更新の時間刻み、電荷密度混合比、バンド幅など)を変えて再度試
してみましたが、症状が一向に変わりませんでした。
この計算では、2種類の原子から成る系を計算していたのですが、これを1
種類のより分かりやすいものに替えて、比較のための計算を行なってみました。
これはデバッグの仕方ページの項目4に該
当します。結果として、1種類での計算は破綻せずに、うまくいく(少なくと
も4、5回程度での全エネルギー発散はない)ことが判明しました。
そこで、再度入力データ(擬ポテンシャル、入力座標、配列宣言部分、ファ
イル入出力指定箇所など)を調べ直しました。そして、見つけてしまいました。
擬ポテンシャルデータに問題があったことが判明しました。
この擬ポテンシャルデータは、前の計算で使い回していた擬ポテンシャルデー
タに新たに必要な擬ポテンシャルのデータを付加して再使用していました。筆
者は元のデータには1種類の擬ポテンシャルデータしか入っていないと思って
いました。ところが実際は、元のデータには2種類のデータが格納されていま
した。それでも前の計算では、最初の1種類目のデータしか読み込まないよう
になっていたので(1種類の系の計算をしている、そのまた前の計算で2種類
の系での計算をしていました)、その段階では2種類分あることを認識してい
たのですが、そのままにしていました。そして今回、新たに擬ポテンシャルデー
タを付加する時には、すっかりそのことを忘れていました。このため、新たな
計算では、2種類めの擬ポテンシャルデータは新たに付加した正しいものでは
なく、元からあった全然異なる擬ポテンシャルデータが読み込まれてバンド計
算が行なわれてしまった訳です。
これでは、計算がうまく行くはずがありません。
本日の教訓は、”入力データのチェックはしつこいほどちゃんとしよう。”
です。
(A)本日のバグ(レポート11/2、1998)
[目次]
今回のバグ(と言うより失敗、間違いの類)も単純です。
表面系の計算で、格子振動の計算をする必要があり、初めて格子振動(フォ
ノン)の計算をしてみたところ、いきなり失敗してしまいました。
原因は、格子振動を求めるために与えた、原子位置のずれのオーダーを間違
えたためでした。
筆者は、以下の入力データの座標部分の元データを、
1000 0.9500 0.1000D-07 6.5000 0
65.8112000000 0.0000000000 0.0000000000
0.0000000000 5.8169400000 0.0000000000
0.0000000000 0.0000000000 5.8169400000
0 COORDINATES 0:NORMALIZED 1:CARTESIAN
0.0061633466 0.0000000000 0.0000000000
0.0663263417 0.5000000000 0.5000000000
0.1273274737 0.0000000000 0.0000000000
0.1885776509 0.5000000000 0.5000000000
0.2499996946 0.0000000000 0.0000000000
0.3114217229 0.5000000000 0.5000000000
0.3726719932 0.0000000000 0.0000000000
0.4336731733 0.5000000000 0.5000000000
0.4938364117 0.0000000000 0.0000000000
0.0022391446 0.5000000000 0.5000000000
0.0652380151 0.0000000000 0.0000000000
0.1266380629 0.5000000000 0.5000000000
0.1883738854 0.0000000000 0.0000000000
0.2499996696 0.5000000000 0.5000000000
0.3116255713 0.0000000000 0.0000000000
0.3733614129 0.5000000000 0.5000000000
0.4347616726 0.0000000000 0.0000000000
0.4977604284 0.5000000000 0.5000000000
次のように変更しました。
1000 0.9500 0.1000D-07 6.5000 0
65.8112000000 0.0000000000 0.0000000000
0.0000000000 5.8169400000 0.0000000000
0.0000000000 0.0000000000 5.8169400000
0 COORDINATES 0:NORMALIZED 1:CARTESIAN
0.0061633466 0.0000000000 0.0000000000
0.0663263417 0.5000000000 0.5000000000
0.1273274737 0.0000000000 0.0000000000
0.1885776509 0.5000000000 0.5000000000
0.2499996946 0.0000000000 0.0000000000
0.3114217229 0.5000000000 0.5000000000
0.3726719932 0.0000000000 0.0000000000
0.4336731733 0.5000000000 0.5000000000
0.4938364117 0.0000000000 0.0000000000
0.0122391446 0.5000000000 0.5000000000
0.0652380151 0.0000000000 0.0000000000
0.1266380629 0.5000000000 0.5000000000
0.1883738854 0.0000000000 0.0000000000
0.2499996696 0.5000000000 0.5000000000
0.3116255713 0.0000000000 0.0000000000
0.3733614129 0.5000000000 0.5000000000
0.4347616726 0.0000000000 0.0000000000
0.4877604284 0.5000000000 0.5000000000
赤色で示したデータの値を0.01ずらし(た値は深紅色で表示)ました。これ
はスラブモデルにおいて、表、裏の各表面のトップレイヤーの原子を構造最適
化で得られた平衡位置からずらしていることを意味しています。そして、この
段階で大きな間違いを犯していました。ここでの0.01を筆者は十分に小さなず
れと思っていました。しかし実際は全然小さなものではありませんでした。
因みに格子振動の計算のための参考論文として、M. T. Yin and M. L.
Cohen, Phys. Rev B26, 3259(1982)を挙げておきます(特に3264
頁)。そこには原子位置のずれの大きさは、0.01から0.1Aとありました。筆者
は、先の0.01のずれを、0.01 a.u.(大体0.005A)と誤認してしまいました。
これは重大な過ちで、ここでの0.01は、表面のスーパーセルの長さ65.8112
a.u.に対する0.01だったのです。つまり実際のずれは、0.658112 a.u.(大体
0.35A)にもなっていました。更に筆者は、同じようにして0.02ずらした場合
(0.7Aものずれ)の計算を行なっていました。いくつかの表面系の計算をして
いて、系によっては0.02ずらすと、計算が破綻(収束しない)している場合も
ありました(この破綻をみて、どうもおかいしと思って、よくよく考えると気
付いた次第です)。
今日の教訓は、”データ入力は慎重に行ない、十分その意味するところも把
握し、何度も検討(検証)ておくべきである。”です。
格子振動の計算もまだ初めたばかりで、正直まだ本当にこれで正しく求めら
れかも、今後検証していかなければならない。
(9)本日のバグ(レポート8/4、1998)[目次]
今回はバグではなく、単なるミス(失敗)です。
筆者は単純な入力データの間違いで、これまで計算した大事な結果の1/3
を台無しにしてしまいました。
(経緯)
遷移金属の計算で、PCC版の擬ポテ
ンシャルの多くにゴーストバンドがあることが判明、これを修正すべく新たに
PCCを考慮した遷移金属擬ポテンしゃルを作成しました。ほとんどのものは、
局所部分(Vcore(r))の修正だけで済みました。これは遷移金属ではs、p、
dを非局所とし、Vcore(r)(BHS論文
で定義されている通りのもの)を記述するパラメーターを、例えば
1.0,0.5,0.5だったのを0.5,0.5,0.5にするものでした。
これを遷移金属炭化物表面の計算をしている途中で気が付き、擬ポテンシャ
ルを修正、バルクの計算までは正しく修正したデータを使用していたのですが、
肝心な表面計算用の入力データを修正するのを忘れてしまいました。実は明日
(8/5)セミナーがあり、この遷移金属炭化物表面の話をするのですが、完
全に真っ青な状態です。
間違いは、0.5とすべき数値を、元の1.0(旧ポテンシャル用)のままで計算
してしまったことです。普通、入力データを間違えると、計算が破綻したり、
収束しなかったり、明らかに間違った結果が得られるのですが、今回はもっと
もらしい結果が出てきたので、かなりの時間問題に気付きませんでした(間違
いもあまりに単純だったこともある)。気付いたときは正に「後の祭り」状態
でした(放心状態)。ただ、後になって結果を良く見てみると確かに若干変な
ところがありました(事前に気付けるほどではない)。
今日の教訓は、「データはくどいほどチェックすべきである。」「誤り、間
違いは単純なほど気付きにくく、そして致命的。」です。
(補足、追加、8/17、1998)
この一件に関して、住友電工の澤村先生から指摘(
感謝)がありました。それは、擬ポテンシャル内に、その元素名、価
電子数、作成時のパラメータ(切断半径、Vcore(r)のパラメータ)及び作成方
法、条件の簡単な説明、使っている単位系、などなどの情報を埋め込んでおけ
ば良いというものです。
こうすれば、バンド計算時に、バンド計算として設定した計算条件と、擬ポ
テンシャル内の内部情報を比較してチェックするようにしておけば、もし互い
の情報に矛盾があれば、すぐに修正して計算をやり直せます(計算の正確性が
増す)。これは単純な間違いや、一体何の計算をしているか分からなくなる事
態を回避するために、大変有効だと考えられます。
(更に補足、8/18、1998)
何と、まだ間違いがありました。それは価電子数の指定のところで、例えば、
Nbは5価(4dに4電子、5sに1電子)として擬ポテンシャルを作ったのに、
バンド計算では4価として計算しているというものです(それでも計算がちゃ
んと収束しているから怖い)。これも大変単純な間違いであり、改めて、上記
擬ポテンシャルの内部情報埋め込みの必要性を認識する次第です。
(またまた補足、9/3、1998)
上記の「更に補足」に関連して、各原子の価電子数が分かると、計算すべき
系の総電子数が分かります。スピンを考えなければ、1バンドに2個の電子が
入り(詰められ)、バンド計算において必要なバンド数が大体分かります。この大体
の意味ですが、バンド数が必要な数より少ないと、計算は普通破綻し
てしまいますが、ではどのくらい余計にとっておく必要があるかが問題になり
ます。
ダイヤモンド(バルク、ダイヤモンド構造でユニットセル内部に4個の場合)
のようにギャップの大きな非金属系では、扱うバンド数=系の全電子数/2と
しても良いのですが、これは非常に限定された話で、大抵の(特に金属的な)
系では、扱うバンド数=全電子数/2という訳にはいかないです。
系が金属的なバンド構造になる場合、ある程度多めにバンドをとっておく必
要があります。例えば、Ta(5価)、C(4価)がそれぞれ9個づつから成る
系(表面系)では、合計で81個の電子が存在し、最低でも41バンドが必要
となります。
この場合、全電子数が81なので、考えるまでもなく系は金属的にならざる
おえなくなっています。では、それぞれ10個ずつで90電子としたらどうな
るのでしょうか?。これは即答はできません。計算してみないことには分かり
ません。これは難しい問題で、本当にこの表面系が金属的になっているかどう
かは、Ta、Cを9個づつでは判定できません。検討の必
要あり。ただし、TiとCの同じ表面系では、Tiは4価なので、全電子数
は72個となり、全てのバンドは詰まっていることになりますが、構造最適化
した場合のこの系の電子状態は金属的となります。
実際の計算では、41バンドでは不足で、より上のバンドまで考慮する必要
があります。ではどこまでとる必要があるかですが。これはあまりはっきりし
ません。以前、扱う系によっては大分上の方のバンドまで考慮に入れないとい
けないという論文(詳細は失念、いわゆるカー・パリネロ的な電子状態計算過
程で必要なのか、部分対角化の時に特に必要であるのかも不明)を目にしたこ
とがあります。筆者も、経験的にバンドは、
十分に余裕をもって多めに取る必要があると判断しています。どのくらいかと
いうと、これも場合場合によるのですが、先のTaとCから成る系(表面系)で
は、 どうも44バンド程度では駄目(どうも力の計算がおかしくなる)で、
51バンド位は必要なようです。但し、45、46、47、、、と全ての場合
でチェックしていないので、どこからが安全、不安の境界であるのかは不明で
す。また系が金属的か絶縁体か、バルクか表面かなどによっても、バンド数の
取り方は変わってしまいます(どこが第一原理な
のだろう^^;)。
従って、事前に擬ポテンシャルに価電子数の情報を盛り込んでおけば、実際
のバンド計算で(最低)必要なバンド数がわかる訳で、バンド計算時にバンド
数に問題があれば(少ない過ぎる、足りないかもしれないなどと)警告するよ
うにできます(更に、実際は状況によりますが上記のような理由により、ある
程度多めのバンド数で計算を行なう必要があります)。
(またまた補足、8/20、1998)
擬ポテンシャルデータ内に埋め込む情報について列挙してみましょう。
- 元素名
- 切断半径
- 原子での電子配置
- 価電子数
- 擬ポテンシャルの種類(BHS, TM, Vanderbilt, etc)
- 作成日
- 作成者
- 作成場所
- LDAの型(Wigner, Perdew-Zunger, MJW, 相対論〔考慮、非考慮〕、
etc)
- GGAを考慮しているか(考慮している場合の型)
- PCC(部分内殻補正)を考慮しているか
- どこまでを価電子と考えているか(価電子数と連動)
- 参照エネルギーの数(普通は一つ)
- バンド計算時に(最低)必要なエネルギーカットオフ(※)
- バンド計算時の計算状況(収束しやすいか、時間刻みの上限等)(※)
- 可能ならば、原子の計算での全エネルギー
- 原子の計算でのスピンポラリゼーションエネルギー
- スピン起動相互作用対応かどうか
- その他(注意事項など)
(※)この情報は、少なくとも一度はバンド計算を行なって、確かめておく必
要があります。(おそらく一度の計算くらいでは、確実なデータは出てこない
ので、ある程度の計算経験を経てから付加できるようにしておくと良いと思わ
れます。)
(8)本日のバグ(レポート3/17、1998)
[目次]
今回のバグも単純です。
プログラムの拡張を行なっていて、ヨウ素のfcc構造でのテスト計算を行なっ
ていて、予想された結果が得られず(格子定数がおかしい)思案していました。
行なった拡張部分は、最初、
C PBE
RGGAEX(I)= (EXPBE + ECPBE)
VGGAEX(I)= (VXUPPBE + VCUPPBE) + (VXDNPBE + VCDNPBE)
というものでした。もうこれを見て何を拡張しようとしているかお分かりの
方々もいるかと思います。
そうです、J. P. Perdew先生のPBEコードの導入を行なおうとしていた訳
です。PBEの詳細は参考文献を見て
ください。また、Rutgers大学のKieron Burke先生のサイト[ページ]からPBEソー
スコードの入手が可能です(取り扱いについては、付随するドキュメント等の
使用上の注意、指示に従って下さい)。
で、上のソースリスト部分は、PBEで計算されたLDA+GGA(PBE)
の結果(EXPBE:交換エネルギー、ECPBE:相関エネルギー、VXUPPBE:スピン
アップ交換ポテンシャル、VCUPPBE:スピンアップ相関ポテンシャル、VXDNPBE:
スピンダウン交換ポテンシャル、VCDNPBE:スピンダウン相関ポテンシャル)
を、こちらのプログラム側で対応する変数(RGGAEX:交換相関エネルギー、
VGGAEX:交換相関ポテンシャル)に代入しています。
この場合、PBEは最初からスピンがアップ、ダウンの別を考慮した形になっ
ていたのですが、こちらの計算では取り敢えず最初はパラの状態で考えること
にしました。つまりスピンアップ側の電荷密度=スピンダウン側の電荷密度=
全体の電荷密度/2として、計算を行ないました。いろいろ曲折はあったもの
の、どうにかうまくいっているのではないかと思えるところまできたのですが、
どうしてもPBE(LDAにしても)の計算での格子定数が正しく出ませんで
した。
どうしようもないと思っていたのですが、同僚の新井さんに相談してみると
いとも簡単に問題点を指摘されてしまいました。
問題は、ポテンシャル代入部分、
VGGAEX(I)= (VXUPPBE + VCUPPBE) + (VXDNPBE + VCDNPBE)
で、アップとダウン両方足し込んでは駄目だと言われました。つまり、
VGGAEX(I)= (VXUPPBE + VCUPPBE)
または、
VGGAEX(I)= (VXDNPBE + VCDNPBE)
だけ(どちらか一方)で良いということです。これは、全電荷密度をP、アッ
プ側をPU、ダウン側をPDとして、パラ(常磁性)では、
P = PU + PD
PU = PD = P/2
V(up) = V(down) = V
V(up)*PU + V(down)*PD = [V(up)*P + V(down)*P)]/2
= 0.5*2*V*P = V*P = V(up)*P = V(down)*P
となるため、スピンを考えない時の交換相関ポテンシャルは、アップ側、ダ
ウン側の一方だけで良いということになります。
ここでの問題は、筆者の知識(勉強)不足であり、教訓はデバッグの常套手段にもあるように、人に相談(議論)す
ることはデバッグにとって非常に重要であるということです(新井さんに感謝)。
GGA(PBE)に関しては、[拡]張編にて
おいおい話ていく予定です。
(7)本日のバグ(レポート1/20、1998)
[目次]
今回はプログラム上のバグではなく、研究上の失敗です。
Pdの擬ポテンシャルは現在、格子定数が実験値より2.6%も長くなってしまっ
ています。これを改善しようとして、切断半径や原子での電子配置などを変更
して、新たにPdの擬ポテンシャルを構築してみました。
そして格子定数が3.89Aと実験値とどんピシャリな擬ポテンシャルを作成す
ることに成功しました。バンド構造(pngファ
イル、9KB)も表示してみてゴーストバンドは存在しないように見えました。
ただちょっと一番上のバンドが平たいなとは思い、ちょっと引っかかるもの
が残りました。しかし、この時格子定数が思った以上に良く出ていたことに気
を取られ、これのもつもっと深い問題を見逃してしまいました。
そして新しいPdの擬ポテンシャルができ、これを今度配布するCD-Rにも収録しようとしてましたが、どうも
バンド構造をみる毎に、一番上の部分的に平たくなっているバンドが気になり、
一応もう少し調べてみることにしました。
そして、より上のバンド(pngファイル、
9KB)まで描いてみました。困ったことに出てきてしまったのです。ゴースト
バンドが、、、
呆れるほどはっきりとした、(奇麗な?)ゴーストバンドがフェルミエネル
ギーの上5eV付近にあるじゃないですか(ショック!)。前のバンドは、6バンド分しか表示しなかったため、
一番上の6バンド目が丁度ゴーストになっているのですが、これは上の表示し
ていない7番目のバンドと重なっていて、平たい明らかにゴーストと分かる部
分を、その7番目のバンドが担っていました。そのため、6番目の一部のみが
平たくなっているだけで、最初このバンドの持っている致命的な問題に気付き
ませんでした。
更に、ゴーストバンドがフェルミエネルギーより上だったのが発見を遅らせ
ました。フェルミエネルギーより上だったため、あまり(と言うよりほとんど)
格子定数に悪い影響を及ぼしませんでした。ゴーストさえなければこの擬ポテ
ンシャルは非常に良いものであったかもしれません。
ゴーストバンドがフェルミエネルギーよりずっと上(数Ry以上)に出てく
るのなら全く問題無しとしてしまうこともできますが、5eVでは小さ過ぎます。
単独ならば良いかもしれませんが、化合物や表面(とそれに原子が吸着する問
題なら尚更)系ではゴーストの存在による悪影響が顕在化する可能性が高いで
す。従って、この新しい擬ポテンシャルは使えません。
今日の教訓は、非常に良い(都合の良い)結果だけに着目して、悪い部分や、
より厳密なチェックを怠る(ポテンシャル作成時にログデル位は見ておくべき
だった。ただみなくても7バンド分表示させればゴーストの存在は明白だった)
ことは、研究上非常に危険であるということです。
因みにゴーストの無い正しいバンド
構造(pngファイル、9KB)を示します。これも6バンド分しか表示してい
ませんが、表示している中で一番上のバンドは、ゴーストのある場合と異なり、
両端が平たくなっていません。当然、その上にもゴーストバンドは(ずっと上
は不明ですが)ありません(但し、この場合は格子定数が良くない)。
(6)本日のバグ(レポート12/5、1997)
[目次]
今回のバグも教訓的です。
あるプログラム(自作ではなく、公開されている他の人のもの)をDEC上で
動かそうとして、コンパイルは全く問題なく終了したのに、いきなり実行させ
ると、
forrtl: error: floating divide by zero IOT trap
で止まってしまいました。メッセージから筆者は単純なゼロ割りで止まって
いるのだと判断しました(実は違った)。
但し、これは自分が作ったプログラムではなかったので、一体どこの箇所で
止まっているのか即座には見当できませんでした。更に、DECのマシン上では、
この時エラーで止まった箇所が示されていません。デバッガーにかければエラー
箇所が分かるだろうと思ったのですが、f77 -g でコンパイルし、デバッガー
にかけると、何か訳の分からないメッセージが沢山出るのですが、肝心なエラー
箇所の表示がなく、この段階で筆者は大変困ってしまいました。デバッガー
(gdb)が表示したメッセージは、
Program received signal SIGFPE, Arithmetic exception.
0x3ff808404f4 in __dpml_signal () at ref_cp/alpha_osf_exception.c:317
ref_cp/alpha_osf_exception.c:317: No such file or directory.
(gdb)
ですが、筆者には何を言っているのかさっぱり分かりません。そこで、デバッ
ガーによる問題解決の道を諦め、別の手で行くことにしました。まずデバッグ
に関する常套手段を基本路線とします。但
し、これは人の作ったプログラムで、この場合1、の正しい結果を与える標準
的な状況が存在しません(これはテスト用のプログラムで、入力データはプロ
グラム内部で全て設定するようになっており、その場合の正しい出力結果に関
しての情報は分かっていました)。
デバッガーが駄目で、コンパイラのオプションを変えても駄目なので、やは
り、プログラム内に多数のチェックポイント(WRITE文)を置いて、しらみ潰
しに探すしかないと思っていたのですが、その前に6、の他のシステムで試し
てみようと思い、他のワークステーション(SUN、HPなど)で試してみたとこ
ろ、SUNやHPでは正しく動作することが分か
りました。ただ正しく動く場合、何のエラーメッセージも出てこないので、こ
れではDECの場合のエラーは、どうして、そしてどこで起こっているのかわか
りません。ただ、これは言語仕様、コンパイラの仕様、実行時の判断の細かい
ところの差によるのではないかと推定できました。
更に思案する内にSUNのマシンにはSUN標準のフォートランコンパイラと、富
士通のフォートラン90コンパイラの二つがインストールしてあり、まだ富士
通の方は試していないので、これを試してみることにしました。そして、SUN
の標準のコンパイラでは正しく稼働したのですが、富士通のコンパイラでは正
しく稼働しないことが判明しました。更に幸運なことにDECの場合とは異なる
エラーメッセージが出てきました。
そのエラーメッセージは、
jwe0266i-e In dx1**dx2 (dx1,dx2:real*8),dx1.eq.0.0 .and. dx2.le.0.0 (dx2=-0.3333333333330000d+00).
と言うもので、エラーの発生したサブルーチンとその発生箇所(何行目か)
もはっきり示されていました。このエラーの意味は、dx1**dx2(dx1をdx2乗)
する時、dx1の値はゼロで、dx2の値が負になっているというものです(富士通
製のコンパイラで、日本語のマニュアルも助けになった)。これは、(もう数
学というより算数は忘れたの自信がないです)1/(dx1**|dx2|)になっていて、
確かにDECのエラーメッセージの言う通りゼロ割りなのですが、正直もっと親
切な表現にして欲しいです(エラー箇所が出てこないのは、やはりおかいしい)。
結局この富士通フォートランでコンパイル、実行した時のエラーメッセージ
で原因はすぐに分かりました。それは、
A = ((1.0D0 + Z1)**Z2 - (1.0D0 - Z1)**Z2) :Z2は負の定数
なっている部分で、Z1が1.0D0または-1.0D0の場合、0**Z2, Z2 < 0という状
況に陥り、エラーになっていることが判明しました。そこで、場合分けして、
Z1が-1.0D0または1.0D0の時にゼロ割りにならないように書き換えると問題な
く稼働するようになりました。
ここでの問題は、SUN、HPでは動いたものが、DEC、富士通のフォートランで
は動かないことです。多分、ゼロ割りの時のエラー処理等の制限が甘い、厳し
いの差と思われるのですが、何とかして欲しいものです。
(5)本日のバグ(レポート10/15、1997)
[目次]
久々に愚かな間違いを犯してしまいました。
前のバグ(4)に関しては、まだ納得していない部分もあり、現在もパラメー
ター文に関しての調査を不定期的に続行中です。
さて、今回のバグは、サブルーチンDIAGON
において、新しい対角化のサブルーチンを導入しようとして遭遇したバグです。
この新しいサブルーチンは、文献「Fortran77による数値計算ソフトウェア」、
渡辺力、名取亮、小国力監修、丸善刊の、エルミート行列対角化ルーチン
EIGCH(当然、これは公開できません)です。
まず筆者が、この書籍に添付されていたフロッピーから、EIGCHを引っ張っ
てきて、計算プログラム本体revpe_d.fにくっつけ、DIAGON
側でCALLするようにしました。
ところが、計算されて出てくる値(全エネルギー)は、全く正しくないもの
でした。
原因は、サブルーチンのCALL EIGCH(----)で使っていた、引き渡し用の変数
の設定を間違ったためでした。
このサブルーチンの正しいCALLは以下のようになります。
CALL EIGCH(ZZZ,IIBA,KNG11,-KGB,KGB,EPS,WWE,LW,EG,ZVN,ICON) 正
ところが最初、筆者は、次のようにして実行していました。
CALL EIGCH(ZZZ,IIBA,IIBA,-IIBA,IIBA,EPS,WWE,LW,EG,ZVN,ICON) 誤
ここで、ZZZが対角化すべき配列(エルミート行列)、IIBAは平面波数(k
点毎に異なる値になる)、EPSは打ちきり誤差、WWE、LWは作業用配列、EGがエ
ネルギー固有値用配列、ZVNが固有ベクトル(波動関数)用の配列、ICONがエ
ラーコードとなります。
間違いは、3番目の変数IIBAです。文献の中での説明には、ちゃんと配列
ZZZの行数とあります。そこを筆者は何を間違
えたのか、可変な変数IIBA(k点に関するループ内で値が変わる)を引き渡し
てしまっています。このため、配列のメモリーか何かが壊れた(?)ため、全
エネルギーがおかしくなったと考えられます。
この3番目の変数をIIBAから、正しい定義による変数KNG11にすると、ちゃ
んと正しい結果を与えることがわかりました(4、5番目の変数はIIBAでも
KGBでも大勢に影響ありません。但し、配列の定義部分も連動して変えておく
必要があります)。
今日の教訓としては、サブルーチンで引き渡している変数の定義、説明はちゃんと読んで、理解しておかなけれ
ばならないことです。変な思い込みで、適当なことをやるとろくなこ
とになりません。
筆者は、このバグに気付くのに3日もかかりまし
た^^;;)。
(4)本日のバグ(レポート6/18、1997)
[目次]
これはこれまでとは、ちょっと趣を異にしたバグです。
最近、新しいDECのサーバーマシンが導入され、最新のOSと最新のフォー
トランコンパイラがインストールされていました。バージョンはそれぞれ、OS
が4.0A、フォートランコンパイラ(FORTRAN77仕様)が4.0でした。
ところが、自分のプログラムが、このバージョン4.0のフォートランコンパ
イラでは、正常に動作しないことが分かりました。コンパイルは無事に成功し、
エラー表示も警告以上のものは出てきません(エラーの出方そのものも、従来
正しく動いておた場合と全く同じものでした)。
そして、実行用オブジェクトを実行させると、思いもしないところで止まっ
てしまいました。エラーはsegmentation faultと
いうもので、最も何を言っているのかさっぱりわからないエラー表示の代表格
と言えるものです。このエラー表示が出たときは、メモリ領域を壊して(何ら
かの不正を行なっている)いることが経験的に多いです。メモリーを壊すのは、
大抵配列の領域外を参照しようとした場合など、配列に関わる扱いのところで
壊し易いです。
しかし、この場合は従来のバージョン3.8以前では、このようなエラーでプ
ログラムが停止することは全くありませんでした。更にDEC以外の機種でも
一応問題なく動いています(富士通VPP、富士通PC用フォートラン、HP、
NECのSX4など)。では原因は一体何なのでしょうか?、DEC側のコン
パイラのバグも疑ってみましたが、問題はやはり筆者のプログラムにあること
が(DEC側の指摘により)わかりました。
問題は、パラメーター文で、設定(定義)した変数を、サブルーチンの引数
として渡しており、引き渡されたサブルーチン側では、変数の名前が変わって
いて、更に、それに値を代入して演算を行なっていました。DEC側によると、
新しいコンパイラーでは、チェックが厳しくなっており、このように実質的に
パラメーター文に代入を行なっているような場合には、エラーとして検出され
るようになり、segmentation faultエラーで、プログラムが止まったと言われ
ました。
この状況を分かりやすくプログラム的に示すと以下の
ようになります。
MAIN PROGRAM
IMPLICIT REAL(A-H,O-Z)
PARAMETER (KNVX=8)
-------
CALL SUB1(KNVX)
-------
STOP
END
C
SUBROUTINE SUB1(NKX)
IMPLICIT REAL(A-H,O-Z)
-------
CALL SUB2(NKX)
-------
END
C
SUBROUTINE SUB2(NKX)
IMPLICIT REAL(A-H,O-Z)
-------
NKX = NKXH + NKXH <--- エラー
-------
END
確かに、この場合おお元はパラメーター文で定義されています。但し、この
変数はサブルーチンに渡される段階で、変数名が変えられています(そして、
そのサブルーチン上でパラメーター文で定義されている訳ではない)。本当に
厳密なFORTRAN77の仕様が、このような扱いを禁止しているのか、少々疑問も
あるので、現在これに関して調査中です。
(8/14)確かに、パラメーター文自体は、
一度定義した後に、定義し直すことができません。つまり、ルーチン内で、定
義されたパラメーター変数に値を代入したりすることはできません。しかし、
サブルーチンの仮引数として、サブルーチンに渡されたあとまでの、厳密な定
義はあるのでしょうか?(--->識者の人々)
ここでの場合、サブルーチンに引き渡されたパラメーター変数は、そのサブ
ルーチン内で、普通の(整数)変数として再定義されていると解釈できないで
しょうか。そうなると、これはエラーではないような気がするのですが?(尚
も、調査中)
DECのフォートランコンパイラも古いバージョンでは、このエラーは引っ
かからなかったのですが、新しいコンパイラではエラーになるようになってし
まいました。DEC側の言い分としては、コンパイラの解釈がより厳密になっ
たとしていますが、ユーザー側からみると(この解
釈が正しいとしても) 「ありがた迷惑」
のような気がします。
このエラーに対する回避手段として、以下のものが考えられます。
MAIN PROGRAM
IMPLICIT REAL(A-H,O-Z)
PARAMETER (KNVX=8)
-------
CALL SUB1(KNVX)
-------
STOP
END
C
SUBROUTINE SUB1(NKX)
IMPLICIT REAL(A-H,O-Z)
-------
CALL SUB2(NKX)
-------
END
C
SUBROUTINE SUB2(NNKX)
IMPLICIT REAL(A-H,O-Z)
-------
NKX = NNKX <--- NNKXには代入などの演算は行なわない
NKX = NKXH + NKXH
-------
END
これは、DECの最新のフォートランコンパイラでも正しく動作することを
確認しました。
(注意)但し、現在(1/26、1999)のD
EC(今はCOMPAQ)フォートランコンパイラでは、上記変更では正しく
動かないです。
或いは、
MAIN PROGRAM
IMPLICIT REAL(A-H,O-Z)
PARAMETER (KNVX=8)
NKX = KNVX <--- 変更点
-------
CALL SUB1(NKX)
-------
STOP
END
C
SUBROUTINE SUB1(NKX)
IMPLICIT REAL(A-H,O-Z)
-------
CALL SUB2(NKX)
-------
END
C
SUBROUTINE SUB2(NKX)
IMPLICIT REAL(A-H,O-Z)
-------
NKX = NKXH + NKXH
-------
END
でも問題ないはずです(これはまだ動作確認をしていません)。
現在(10/31)この方法を試してみたところ、テスト用の小規模な計算
ですが、うまくいくことを確認しました。
現在(1/26、1999)でも正しく動くことを確認。
ただし新たな問題が出てきました(10/31)。
まず、このバグに対する最初の処方では、DEC上のコンパイルとして、
f77 -r8 -O5 sample.f
では正しい結果を与えますが、
f77 -r8 -O4 sample.f
では、実行中segmentation faultエラーで
止まってしまうことが判明しました。コンパイル時にはこれに関係しそうなエ
ラーメッセージは出てきません。また、最適化オプション-O2,-O3,-O4の場合はsegmentation faultエラーで止
まりますが、オプション-O0,-O1,-O5の場合
は正しく動くことが確認できました。ここで、注目されるのは、最も最適化し
た場合の、-O5では正しく動いてしまうこと
です(普通、最適化の度合いを上げれば、エラーで止まることが多くなるのに、
今回はその逆になっています)。
この場合、今後も-O5のオプションによる最適化状態で、より実際的な計算
を継続していって良いものかどうか不安です(まだ試していませんが、より大
規模な計算になると-O5オプションでは動かなくなる可能性があります)。当
然、より古いバージョンのDECフォートランコンパイラではこのようなこと
は起きません。
同じく、-g0,-g1,-g2(デバッグモードに関してのオプション)では正しく
動きますが、-g3オプションでは動きません。動かない場合に対して、デバッ
ガーを使ってみましたが、訳のわからない(どうみてもエラーでない)箇所で
止まっています。どうやら、メモリー領域か何かを壊していて、見当違いなと
ころでエラーになっている(バグとしては最悪のパター
ン)ようです。
後の解決方法(処方)でも事情は全く同じで、-O5では動きますが、-O4では
動きません。
どうもやっぱり、DECの新しいバージョンのコンパイラの方にも何か問題
がありそうで、本当の原因究明と根本的な解決のための調査が必要(でも時間がない)と考えられます。
正直な話(2/9、1998現在)、次のコンパイラのバージョンアップで
の改善を期待していた(今までうっちゃってあった^^;)のですが、DECが
何とコンパックに身売りすることになってしまいました。Degital UNIXの将来
に一抹の不安を憶えます。
(6/30、1998)現在、DECのフォートランコンパイラのver4.0で
駄目でしたが、最近、最新のバージョンver5.0でもやはり駄目であることが判
明しました。どうもコンパイラの仕様そのものが、上記のような記述を認めないという方針になってしまったようです。せ
めてオプションで回避できるようにしてくれたらと思う次第です。
実は、DEC側に別に最新である必要もなかったので、OSはそのままで、
フォートランコンパイラのバージョンを従来のプログラムで問題無く動く
ver.3.8にしてもらいました(これも前向きではないですが解決策の一つ)。
このバグの問題は、もともとパラメーター文で定義されていた変数に対して、
設定変更(代入)を行なおうとしたためなのですが、これにはサブルーチン
(SUB1、SUB2)が後から付け足されたもので、もともとメインプログラムと整
合性があまりなかったことにあります。それをプログラムの拡張のために無理
にくっつけた感があります。改めて、プログラムの設計や拡張は十分に練って
行なう必要があることを認識しました(そうでないと、後々面倒になる場合が
あることを思い知る)。
(3)本日のバグ(レポート3/26、1997)
[目次]
最近、プログラム開発がなかったので、あまりバグも出てこなかったのです
が、とうとうバグ(というよりプログラムの構造、仕様上の問題)を見つけて
しまいました。
まず問題となった部分(計算結果)を以下に示します。
TOTAL SUMM OP1 = 5.930788159663123e-04
2 = 0.0000000000000000e+00
3 = 0.0000000000000000e+00
4 = -1.251707515445920e-03
5 = 0.0000000000000000e+00
6 = 8.736301364726182e-04
これは正方格子におけるGaのストレスの値です。1番目、4番目、6番目の
値がそれぞれx方向、y方向、z方向に対応しています。この値を見てすぐに
おかしいと思ったら、貴方はするどいです。
扱った系が正方格子(正確には体心正方格子、bct)なので、明らかに上
の3つのストレスの値の2つは値が同じでなければなりません。ところが3つ
とも異なる値になっています。明らかにこの計算結果は間違っています。
この間違いは、ある条件設定の場合に起こります。そうでない場合は、以下
のように正しい値が出てきます。
TOTAL SUMM OP1 = -3.386523218593673E-003
2 = 0.000000000000000E+000
3 = 0.000000000000000E+000
4 = -3.430875185404469E-003
5 = 0.000000000000000E+000
6 = -3.430875185401056E-003
対称性から、y方向の値とz方向の値が(計算誤差を除いて)一致していま
す。では、どのような条件の時に値がおかしくなるかと言うと、ストレスを使
ってユニットセルの形についての構造最適化を行なう場合であることがわかり
ました。単に、電子状態のみの計算で、最後にストレスの計算を行なう場合は
上のように正しい値を返しました。
最初、筆者はこれをVPP上で計算していたので、間違った先入観に因われ、
原因は並列化部分ではないかという方針で、デバッグを行ないましたが、全く
原因を見つけられませんでした。
しかし、土、日(3/22、3/23)と休んでいる時に、家でもう一回正
しく出ている場合と正しく出ていない場合の計算出力結果を比較検討してみる
ことを思い立ちました。そして、昨日(火曜日、3/25)に以下の違いに気
付きました。
間違っている方
---------- THE FERMI ENERGY = 0.9234210201451082d-01 26.000000
CALL FORZFB
REAL TOTAL CHARGE = 25.99999999997447 IN XCFFT
TOTAL ENERGY FOR 100-TH ITERATION=-131.9255525 -0.1319256d+03
ITER=100 ET(H)= 0.2962622d-07 ET(M)= 0.000806 DC= 0.3412940d-06
---------- THE FERMI ENERGY = 0.9234210813638645d-01 26.000000
TOTAL STR 1 1 = -0.3181689275022663
2 = 6.268243606208947e-04
3 = 6.268243606208876e-04
4 = -0.3180186954074477
5 = -6.008948961606797e-05
6 = -0.3180186954074479
正しい方
---------- THE FERMI ENERGY = 0.2661819904309356D+00 26.000000
CALL FORZFB
REAL TOTAL CHARGE = 25.9999999999665 IN XCFFT
TOTAL ENERGY FOR 100-TH ITERATION=-131.8150778 -0.1318151D+03
ITER=100 ET(H)= 0.4234662D-06 ET(M)= 0.011518 DC= 0.5330057D-06
---------- THE FERMI ENERGY = 0.2661826508252645D+00 26.000000
TOTAL STR 1 1 = -0.526384186291203
2 = 1.794706243030863E-003
3 = 1.794706243030874E-003
4 = -0.526410035928503
5 = -4.039471827445321E-005
6 = -0.526410035928507
100回目まで電子状態の計算のみを行ない、101回目にストレスの計算
を行なうのですが、良く見ると、赤色(色の出ない場合は御容赦下さい)で示
した部分、つまりサブルーチンFORCE(またはFORZFB)を呼び出し
ている部分が正しい方と間違っている方で異なることがわかります。
この場合、FORCEを呼び出すのが正しいのです。FORZFBでは正し
いストレスの計算が出来ません。間違った方はこのFORZFBを呼び出し、
ストレスの値が正しくなくなってしまているのです(FORCEでは後で計算
するストレスのサブルーチンで必要な配列変数の計算を行なっているが、FO
RZFBではそれを行なっていないため)。
そして、この違いを生ずる根本的な原因は
メインプログラム部分にあります。
IF (MOD(ITER,IMDI).EQ.IRES) THEN
IF (ITER.EQ.1 .OR. ITER.GT.IMD) THEN
WRITE (6,*) 'CALL FORCE'
CALL FORCE(IREC8)
ELSE
CALL FORZFB
END IF
ELSE
WRITE (6,*) 'CALL FORZFB'
CALL FORZFB
END IF
上の赤い部分(赤くなってない人ごめんなさい^^;)の制御変数、IMDIによ
る条件判定に重大な問題があります。この変数は、原子間に働く力によるユニッ
トセル内の原子の構造の最適化用のもので、ストレス用ではありません。従っ
て20回おきにストレスの計算をするように設定(CONTL.DATで設定)しても、その制御変数
はIOVEであるため、ストレスの計算のために必要なサブルーチンFORCEは
いつまでたっても呼び出されません。これでは正しいストレスの計算はできま
せん。
解決策としては、いまのところ2つ考えられます。(案1)上の条件判定部
分を以下のように変更する。
IF (MOD(ITER,IMDI).EQ.IRES .OR. MOD(ITER,IOVE).EQ.IRES) THEN
この方法はまだ試していませんが、プログラムそのものを修正するので、根
本的な解決策と言えます。一方、(案2)はCONTL.DATを以下のようにします。
0
1.0D0 0.0D0 5.0D0
0.7D0 0.005D0 300.0D0
-0.0034D0
0.0D0
0.0D0
-0.0034D0
0.0D0
-0.0034D0
0 0 2 0 12
100 20 20 1 200 0 200 0
0
実は、このbct-Gaの系は対称性からユニットセル内部の原子は動きません。
従って原子間の働く力を計算する必要がありません。そこで上のデータの最後
から2番目の行を 100 20 1 200 0 200 0としてい
たのです。これでサブルーチンは201回目(実際の計算は200回で終るよ
うになっている)にならないと呼び出されません。そこで、上のように原子間
に働く力とストレスによるユニットセルの最適化も20回毎に計算するように
すれば良い訳です。但し、この系では原子は動かないので、原子のための動力
学用の時間刻みをゼロに設定してあります。
この方法は、プログラムを修正しないで、入力データの変更だけで対応でき
るので、作業が簡単ではありますが、ユニットセル内の原子の最適化操作(刻
み時間をゼロにしたので、原子は動かないし、この系では対称性から力そのも
のが出てこない)を行なうので、若干計算に無駄が生じます。
ここでの重大な問題は、これはバグと言うより、プ
ログラム設計、仕様上の重大な欠陥であり、これをこのプログラムを作った当
の本人が認識できなかったことにあります(バグが仕様になっているとも、仕
様がバグになっているとも言える)。
まさ
にしようもないプログラムと言えます。
(2)本日のバグ(レポート2/6、1997)[目次]
今回のバグはそう深刻なものではありませんが、よくやるバグです。
プログラムを改良中に、あるDOループ内の配列変数を、他のDOループ内
に移動(複写でも同じようにバグるはず)して、コンパイルは何の問題もなく
終了したのに、いざ実行してみると計算結果が明らかにおかしくなってしまい
ました。
原因は簡単で、移動(または複写)した配列AがDO 100 K=1、10
0というループ内にあったのに、移動(複写)先のDOループの変数がKでは
なくNだったためです。
DO 100 K = 1,100
A(K) = A(K) + 1.0DO
100 CONTINUE
を、
DO 200 N = 1,100
A(K) = A(K) + 1.0D0
200 CONTINUE
としてしまいました。これではちゃんと動きません。ここでの問題は、同じ
繰り返し数で扱うDOループの制御変数(KやN)が統一されていないことで
す。行き当たりばったり的に作ったプログラムは、特にこのような単純な間違
いで足元をすくわれてしまいます。
(自戒の意味を込めて)プログラムを作る時は、同じことをするDOループは
同じ制御変数を使いましょう。
(1)本日のバグ(レポート1/23、1997)
[目次]
新しい計算ルーチン作成中に、今日の作業はここまでと、敢えて注釈を付け
ずに------------------------------を入れておいたのを完全に忘れ、翌日そ
のままコーディング作業を続けてコンパイル、実行してみるとバグの嵐だった。
このプログラムはSUNのOEMマシンだったが、コンパイルエラーメッセージ
だけでは原因が良く分からなかった。ところがDECのマシンでコンパイルす
ると同じ箇所でコンパイルエラーとなるが、エラーメッセージからすぐに、注
釈を付けずに、----------------------------をコード内に挿入していたこと
がわかった。
この問題の重要な点は、筆者がDECが主に使用するマシンで、SUNのマシ
ンはごくたまにしか使わないため、あまりSUN上でのデバッグの経験が乏しい
ことにあります(その前に、ちゃんとマニュアルのコンパイルエラーメッセー
ジの説明を読んでいないのが最大の問題かもしれません^^;)。
既に昨日のことになってしまいましたが、ここで指摘したいのは、コンパイ
ラーのエラーメッセージの意味を良く理解しておくこと(エラーメッセージの
出し方や、メッセージの内容の貧弱さ、難解さにも問題がありますが)、わか
らないメッセージに出合ったら、ちゃんとマニュアルを読むことです。偶然、
異なるシステム同士の結果の比較からエラーの原因を突き止めましたが、これ
はエラーメッセージの意味をちゃんと把握しておけば、そのようなことをする
必要のないバグでした。
[先頭][総目次
][最初に戻る][Top][失敗知識データベース(JST)]