バンド計算プログラム+用語集(拡張編) [Top]
- 目次 [Top]
- はじめに
- ストレス編
- 磁性+交換相関項+局所密度近似編
- GGA編
- スピン軌道相互作用(失敗)編
最初の思惑や、方針とは裏腹に、どんどん変な方向に向かっています。
(10/29、2004)磁性編の前にストレス編を置く。
- ストレス(stress):テンソル量
- 具体的な計算例:サブルーチンSTRNL
。
- 非局所擬ポテンシャルのストレス部分の表式
(pngイメージ[30KB])の計算を行ないます。
- ストレスの計算の判定方法:一例。
- ストレス関連文献
- 部分内殻補正に対するストレスの表式(pdf形式、159 kb、Appendix参照、CMSMD'96の原稿pdf)
ストレス計算に関しての覚書(備忘)。
- Ωρが一定なので、∂(Ωρ)/∂Ω = ∂(Ωρ)/∂ε = 0である。←ρは電荷密度
- ストレスの対角成分が圧力。P = -∂Etot/∂Ω (P:圧力、
Etot:系の全エネルギー、Ω:系の体積←単位胞、スーパーセル
等の体積)
- ストレスそのものはテンソルで表される。
- ↑従って、1/σαβ = (σαβ)-1
は、単なる数値的な割算ではなく、逆行列として考える必要がある。
- σαβ = 1/Ω ∂Etot/∂εαβ
σαβ:ストレス、α,β=(x,y,z)、εαβ:格子
の歪み。ストレスを求める具体的な表式は、以下の文献(特に[2])を参照。
(2/25、2005)マイナス符合が付か
ないことを確認。下のストレスと圧力の関係参照。
- P = - (σxx + σyy + σzz)/3
となる。P、σの定義式の符合に関しては、上記と異なる表記である場合も
散見される(調査中)。
- O. H. Nielsen, R. M. Martinによる以下の2論文は特に必見。
[1]O. H. Nielsen and R. M. Martin, Phys. Rev. B32, 3780(1985)
[2]O. H. Nielsen and R. M. Martin, Phys. Rev. B32, 3792(1985)
- ストレス(または圧力)を使って、セル(単位胞、スーパーセル等)の
構造最適化が可能。一例:First-Principles Variable Cell Shape
Molecular Dynamics(FP-VCS-MD)による方法
- ストレス(圧力)が、直接的に働くのは単位胞(またはスーパーセル)
の面(結晶格子面、或は結晶格子の形状と言ってもよい)。これにより動くの
は結晶格子の面である(間接的には、これにより原子も相対的に動いてはいる)。
- Asymmetricなスラブで生じる双極子モーメントによる表面ストレスへの
補正は、無視出来るほど非常に小さいことが、P. Feibelman,
Phys. Rev. B72, 153408(2005)の論文で示されている。
- 平面波数(←基底関数)に関しての収束は、全エネルギーや力よりも注
意する必要がある。平面波数が少ない(エネルギーカットオフが小さい)場合、
正しいストレスが得られない。
- ↑TM型のノルム保存型擬ポテンシャルでは、シリコンで25Ry以上、
炭素など(周期表第二列)では70〜80Ry以上。遷移金属でも70〜80
Ry以上必要。但し、これは目安であり必要な平面波の数は切断半径の設定値、
ポテンシャルの型、種類などに依存する。↑
- ストレスの単位:バンド計算での単位を参照。→例:100 GPa = 0.0034 a.u. = 100 万気圧 = 1 Mbar
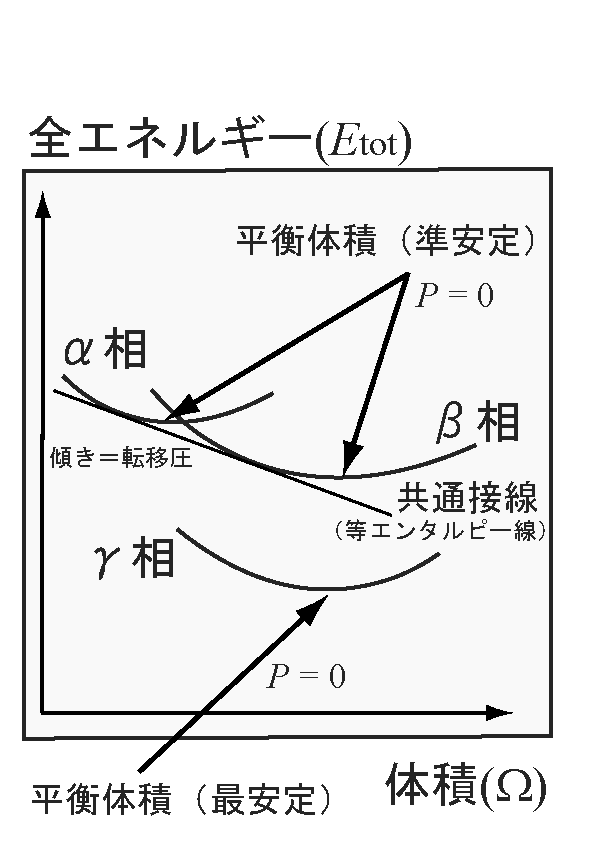
- 圧力誘起による構造相転移の場合、転移前
の相(結晶構造)と転移後の相(結晶構造)における、それぞれの全エネルギー-
体積曲線の共通接線の傾きが転移圧となる。←
縦軸を全エネルギー、横軸を体積とする座標上の曲線の傾きは、P = -∂
Etot/∂Ωから圧力であることが分かる。この時、圧力は静水圧
(等方的圧力)を考えている。
- ↑参考文献↑: A. Mujica, A. Rubio, A. Munoz and R. J. Needs, Rev. Mod. Phys. 75, 863(2003)[Common tangent method]
- 圧力下での構造の安定性に関しては、エンタルピー(H = Etot + PΩ、Hはエンタルピー、Pは圧力)での比較が有効。因みに、エンタルピー(= Enthalpy)である。
- 上記の、共通接線上では転移前の相(相1)と転移後の相(相2)のそれぞれのエンタルピーが一致している。つまり、
H1 = E1 + PΩ1、H2 = E2 + PΩ2
において(圧力Pは相1、相2で共通=等しい)、
P = - ∂E/∂Ω = - (E2 - E1)/(Ω2 - Ω1) = ΔE/ΔΩ
であり(ΔE = E1 - E2 > 0としている)、
H2 = E2 + PΩ2 = E1 + PΩ1 - ΔE + PΔΩ = H1
である。ここで、- ΔE + PΔΩ = 0である。
- ↑参考図(png形式、12 kb):α、β、γ(最安定)の3つの相における、全エネルギー - 体積曲線。α相、β相に共通の接線が等エンタルピー線であり、この線の傾きが転移圧に相当する。注:図とウェブページ上の文章とは必ずしも整合していない。
- エンタルピー関連:圧力が異方的な場合での扱いに関しての参考文献
- [1] C. Cheng, Phys. Rev. B67, 134109(2003)
- [2] C. Cheng, W. H. Huang and H. J. Li, Phys. Rev. B63, 153202(2001)
- [3] M. Cococcioni, F. Mauri, G. Ceder and N. Marzari, Phys. Rev. Lett., Vol. 94, No. 14, 145501(2005)[Electronic-Enthalpy functional][Finite system]
- K. Gaal-Nagy and D. Strauch, Phys. Rev. B73, 134101(2006)[Transition pressure][[Enthalpy barrier][Nonhydrostatic]
- 圧力下での弾性定数の扱い(参考:弾性定数の計算)
- P. M. Marcus, H. Ma and S. L. Qiu, J. Phys.: Condens. Matter 14, L525(2002)
- 圧力に関して興味深い論文
- H. Katzke, U. Bismayer and P. Toledano, Phys. Rev. B73, 134105(2006)[High-pressure structural phase transition][Si, Ge, Sn, Pb]
- 硬さ(固さ、堅さ、hardness)について
- J. Dong, J. K. Tomfohr, O. F. Sankey, K. Leinenweber, M. Somayazulu and P. F. McMillan, Phys. Rev. B62, 14685(2000)[VASP][Hardness][Bonded][Nonmolecular]
- M. Hebbache, Solid State Communications 113, 427(2000)[Shear modulus][Hardness]
- F. Gao, R. Xu and K. Liu, Phys. Rev. B71, 052103(2005)[CASTEP][Origin of hardness][Spinel]
- Z. Ding, S. Zhou and Y. Zhao, Phys. Rev. B70, 184117(2004)[DFT++][FHI98PP][WIEN2k][Hardness][Fracture toughness][Brittle]
- F. Gao, Phys. Rev. B
73, 132104(2006)[Theoretical model][Intrinsic hardness][Mulliken analysis]
- J. S. Tse, D. D. Klug and F. Gao, Phys. Rev. B73, 140102(R)(2006)[VASP][Hardness][Nanocrystalline]
- X. Guo, et. al., Phys. Rev. B73, 104115(2006)[CASTEP][Bond ionicity][Hardness][Structured]
- X. Guo, et. al., Diamond and Related Materials 16, 526(2007)[CASTEP][Hardness]
- S. Veprek, A. S. Argon and R. F. Zhang, Philosophical Magazine Letters, Vol. 87, No. 12, 955(2007)[VASP][Origin of the hardness enhancement][Superhard][Ultrahard][Nanocomposite]
- 【参考文献】星野公三、パリティ物理学コース「固体はなぜ固いか」、丸善株式会社(1996)
- 最近は、LAPW法でのストレス計算も可能になっている(参考文献の(5)、(7)参照)。
- 静水圧なら、系の全エネルギーと体積から、マーナハンの式を使って当該する体積での圧力を求め
ることが出来る。
- 擬ポテンシャル法で、高い圧力下での計算を行なう場合、切断半径及び
内殻からの寄与について注意する必要あり。
- 構造の安定性を議論する上で、フォノン
の振動数が虚数(便宜上、負の振動数と言うことがある)になる場合、それは
原子の平衡位置近傍の全エネルギーの振舞いが調和的でなく、むしろ原子が変
位するとエネルギーが下がることを意味している。つまり平衡位置からずれた
方がより安定となってしまう。→これは平衡位置が安定位置でない(=構造的
に不安定)ことを意味する。(注意:以下の記述〔←間違ってるかもしれない。
従って無保証〕に関して更に調査中、2/13、2009)但し、これだけで
は系そのものが必ず構造相転移することを意味しない。負のフォノン振動数は
元構造での特定の原子の平衡位置が不安定であるこを示すが、ユニットセルや
原子の構造が歪んだり、変位したりして構造相転移した場合(←これにより負
のフォノン振動数が出なくなったとしても)の全エネルギー(又は自由エネル
ギー)が、元の構造より低いことまでは保証していない。
- 構造相転移を扱う場合、全エネルギー、エンタルピー、自由エネルギー
の大小について比較する必要がある。
- 結晶構造は、高圧下でより高い対称性を持つ構造に転移しそうであるが、
必ずしもそうではない。圧力が高くなり原子間が短くなっていけば、系は
配位数(ある原子の周り〔近く〕に何個の原子があるか)をより多くする
ように構造を変化させる(←必ずしもこうなるとは限らない)。これは必ずし
も高い対称性になることを意味しない。例えば、体心立方格子の最近接の原子
数(配位数)は8であるが、これが体心正方格子になると、最近接的
な配位数は10(c軸が縮む場合)または12(a,b軸が縮む場合)となる。
- 非常に高圧では体積が大幅に減少する。例えば、体積が圧力ゼロの場合
と比べて1/10になったとすると、電荷密度は単純な平均として考えれば10倍
になる。但し、rsパラメーターへの効きは、(1/ρ)
1/3なので、体積が1/10になったとしてもrsの値は平
均としておよそ2.15倍小さくなるだけである。但し、更に極端に体積が縮まれ
ばrsの値が、通常の圧力ゼロを想定した局所密度近似の表式で有
効なrs値の範囲外に出る可能性がある。
- rsパラメータ: 4π
(rsaBohr)3/3 = 1/ρと表現すると(ρ:
電子の密度→[電子の個数]/[体積]、 aBohr = 0.529177 A = 1
a.u.)、rsは無単位量となる(ここで[個数]は単位と考えない)。
rsを無単位量とするため、rsでのρは電荷密度ではな
く、電子の(数)密度である(1/29、2010)。
- 高圧下で予想される現象:内殻の価電子化←実験的には観測されていな
い。擬ポテンシャル計算では、内殻電子部分を価電子とみなして作成する必要
がある(例:Gaの浅い3d内殻軌道←
筆者等の研究)。
- 異方的圧縮下での負のポアソン比的挙動が理論的に見つかっている(筆
者等の研
究: LiBCなど)。
- ポアソン比: -1 ≦ σ ≦ 1/2 ← 体積不変の条件から、1/2(上限)、形一定の条件から、-1(下限)が決まる。一様であることが前提。
- 因みに、原子に働く力Fは、F = - ∂Etot/∂Riである。Riはi番目の原子の座標ベクトル。ユニットセル内の原子を動かす(構造最適化或は構造緩和させる←格子自身〔セル〕の緩和とは異なる)には、この力を使う。
- (参考1)エンタルピー(= Enthalpy): H = Etot + PΩ、Hはエンタルピー、Pは圧力。
- (参考2)ギブスの自由エネルギー: G = Etot + PΩ - TS、Tは温度(K)、Sはエントロピー。
- ストレス関連の有用[ページ](アクセス不能を確認。新井氏による)
既にご存知のように本WWWサイトで公開されている第一原理分子動力学計
算用プログラム、revpe_d.fでは、常
磁性状態でのバンド計算しか行なうことができません。
既に強磁性等、スピンが分極化した場合に対応するプログラムは完成してい
ましたが、まだ公開していませんでした。ここでは、この磁性問題対応版プロ
グラムの公開と説明(開発方法)を行なうことを第一の目的として話を進めて
いきたいと思います。
現時点では、磁性状態としては、単純なFe,Co,Niの強磁性状態の計算テスト
しか行なっていません。より複雑な磁気構造(反強磁性、フェリ磁性など)に
関しての計算、経験の蓄積は全くありません。
では実際に現行の、revpe_d.fまたはrevep_d_new.fなどを、磁性を扱えるよ
うにする具体的な方法について、ここでは議論したいと思います。磁性を扱え
るようにするということは、これまで全く考慮していなかったスピンのアップ
とダウンを別々のものとして考えるということです。常磁性のときは1つのバ
ンドに2つの電子(アップとダウン)があると考えましたが、磁性を考える場
合は、1つのバンドには1つの(アップ、ダウンいずれか)が対応するように
なります。
ではプログラムのどの部分を変更する必要があるのでしょうか。
最も重要なのは交換相関項の部分です。これを磁性考慮版に変更する必要が
あります。revpe_d.f等ではWignerの表式
を用いていましたが、残念ながらこの表式では磁性を考慮することができませ
ん(そのような表式が用意されていない)。従って、Wigner以外の交換相関項
形式を採用する必要があります。この場合の詳しい表式に関しては、文献「密
度汎関数法とその応用‐分子・クラスターの電子状態」、菅野 暁監修、里子
允敏、大西楢平著、講談社サイエンティフィック刊、4.5.1節、112頁
周辺に幾つかの表式が載っていて大変参考になります。
本計算では、ノルム保存型の擬ポテンシャルを使用していますが、基本的に
は何ら変更を加える必要はありません。但し、本擬ポテンシャル(NCPS9
7、2K)は、その大半が先のWignerの表式を採用しています。従って、バン
ド計算部分との整合性を考えれば、擬ポテンシャルもバンド計算側に対応する
交換相関形式でのポテンシャルの再構築の必要性があります。ただここで扱う
擬ポテンシャルは、unscreeningしたものを用いるので、擬ポテンシャル側と
バンド計算側の交換相関項の形式が合わなくとも、そう深刻な影響はないと思
われます。
交換相関部分に渡す情報は電荷密度に関するもので、分極(偏極)に関して
のパラメーターζ、
ζ = (ρ↑ - ρ↓)/(ρ↑ + ρ↓) = (ρ↑ - ρ↓)/ρ
ρ = ρ↑ + ρ↓
ρ:全電荷密度、ρ↑:up spin、ρ↓:down spin
及び、rs(ρ)パラメータ(無単位量)。無単位量のため本当は、
rsでのρは電荷密度ではなく、電子の(数)密度である。ハート
レー原子単位では電荷の単位が、e2 = 1(リュードベリ原子単位
では、= 2)なので、暗黙の内に電荷がかかっていると考える。分極に関して
のパラメーターは、常磁性(ρ↑ = ρ↓ = 1/2ρ)での交換相関項の表式と、
スピンが完全に分極した場合(ρ↑ = ρ, ρ↓ = 0またはその逆)の表式を
内挿するために使われる。つまり、交換相関エネルギーεxcはス
ピン分極を考慮した場合、
εxc[ρ,ζ] = εxcparr(ρ) + ( εxcferro(ρ) - εxcparr(ρ) )f(ζ)
εxcparr(ρ):スピン分極していない場合
εxcferro(ρ):スピンが完全に分極している場合
f(ζ) = { (1 + ζ)4/3 + (1 - ζ)4/3 - 2 }/{ 2(21/3 - 1 ) }
という内挿表式が使用される。常磁性(ρ↑ = ρ↓ = 1/2ρ)なら、ζ =
0であり、f(ζ = 0) = 0となる。従って、
εxc[ρ,ζ] = εxcparr(ρ)
となる。一方、完全に分極した場合(ρ↑ = ρ, ρ↓ = 0またはその逆)、
ζ = 1またはζ = -1となり、f(ζ) = 1となる。従って、
εxc[ρ,ζ] = εxcferro(ρ)
となる。それ以外の分極した場合は、parrとferroが上記内挿式にあるよう
に適当に内挿された交換相関エネルギーの値となる。交換相関ポテンシャルも
ほぼ同様な内挿が行なわれる。
アップ、ダウン別に擬ポテンシャルを構築する必要があるのは、スピン軌道
相互作用を考慮する場合で、単に(分極した)磁性を扱う場合は、そのような
ことを考慮する必要はありません。
交換相関項の部分以外に変更を必要とするのは、波動関数に関しての部分で
す。波動関数はアップ用、ダウン用の二つが必要になります。このため、総電
荷密度以外に、アップ用、ダウン用の電荷密度も必要になります。当然、計算
全体でもアップ用の計算、ダウン用の計算の二つに分かれます。このループ
(アップとダウンで2回しか回らない)はバンド計算において、一番外側で、
最大のループといえます(k点のループの外側になります)。
一番簡単で、分かりやすいのは、k点ループの外側に、スピンに関しての
(アップ、ダウン)のループを作ってしまう方法です。
DO 100 ISPIN = 1,IFP+1
Paramagnetic case --> IFP = 0, Ferromagnetic case --> IFP = 1
------
DO 200 IK = 1,KNV
------
WAVE_FUNCTION(IK,ISPIN) = UP or DOWN
------
200 CONTINUE
100 CONTINUE
ループに関しては別の手段もあります。それはk点のループを2倍にして、
それぞれ前半をアップ用、後半をダウン用(逆でも別によい)とする方法です。
DO 100 IK = 1,2*KNV
------
WAVE_FUNCTION( 1, KNV) = UP
WAVE_FUNCTION(KNV+1,2*KNV) = DOWN
------
100 CONTINUE
既存の磁性非考慮のプログラムから、磁性考慮のプログラムへ拡張する場合、
後者の方法が最も簡単に取り掛かることができると想定したのですが、実際に
は前者のスピンに関してのループを新たに導入する方法を選びました。
後者の方法を選ばなかった理由は、最初k点ループ数を2倍にしてアップと
ダウンの計算を割り振ろうとしたのですが、その制御が思った以上に面倒そう
だったので(例えば、常磁性、強磁性の場合分け)、むしろ一番外側にスピン
用のループを作った方が楽だと判断しました。
(8/2、2018)k点のループを2倍にする方法は、並列化(k点並
列)にとっては有利に働くと思われます。系が大きくなると、相対的に必要
なk点数が少くて済むようになり、並列化にとっては不利になります。k点
が2倍ならば、その分並列化による効果が効くので有利に働きます。
(8/17、1998)最近気付いたのですが、k点のループ以外にバンド
のループ数を2倍にする手もあります。バンドはアップスピン用のバンド、ダ
ウンスピン用のバンドと分かれているので、k点を2倍にするよりかは拡張し
易いと思われます(ただ、筆者はこの方法は試していません。正直現在思い付
いた段階なので、思わぬ困難があるかもしれません)。
ただ、良く考えると、バンドに関しての配列変数EKO(KEG,KNV)は、先の前者
の方法でもEKO(KEG,KNV,2)となっています。EKO(2*KEG,KNV)として(k点では
なく)バンド関係のループを2倍にすることに利点があるのかどうか、現在の
ところ不明です(あまりないようにも思えます)。
これら以外の方法としては、スピンアップ部分とダウン部分の計算を独立で
行なってしまう手があります。スピンに関する計算も独立性は高く、ほとんど
の計算部分で(アップ、ダウン)互いに影響を及ぼすことはありません。アッ
プ、ダウン両方の部分が必要なのは、交換相関項の計算部分、フェルミエネル
ギー決定部分と全エネルギー計算部分、力やストレスの計算部分などです(電
子状態を計算するだけなら、力やストレスの計算は必要としません)。力、ス
トレスの部分を除けば、計算ルーチンとしての量及び計算時間として占める割
合は大して多くはありません。従って、バンド計算のほとんどの部分をアップ、
ダウンスピン別々で計算できます。
これは並列計算からみても有利で、アップ、ダウン別々のCPUで並列で計
算させることが可能です(但し、2CPU並列しかできないのが欠点)。FL
APW法のパッケージで有名なWIEN97
(現在は、WIEN2k)は、アップ、ダウ
ンを別々に計算する(場合によっては並列での計算も可能か?)場合のシェル
プログラムが標準で用意されています(いるはず)。
スピンのループを作る場合、DO 100 ISPIN =
1,IFP + 1 とかして、変数IFPを、常磁性のときはIFP = 0、強磁性の
ときはIFP = 1と場合分けする方法が考えられます。このときIFPは、プログラ
ムの最初で指定する(この場合、常磁性、強磁性でいちいち再コンパイルする
必要があります)方法もありますが、筆者は入力データ上(CONTL.DATの磁性
対応版、まだ公開していません)で制御するようにしています。こうすれば再
コンパイルの必要はなくなります(本当は、磁性考慮、非考慮で配列のサイズ
が変わりますが、少々無駄を承知で最初からスピンアップ、ダウン分として配
列を宣言しておけば再コンパイルの必要性はなくなります。当然、大規模な系
ではこの手は使えません)。
実際、スピン用のループを作ると、既存の波動関数(固有ベクトル)や電荷
密度などの変数も、スピン用に書き直す必要が生じます。例えば、固有ベクト
ル用の配列変数、ZAJ(KNG1,KEG,KNV3)は、ZAJ(KNG1,KEG,KNV3,2)となります。他にも電荷密度関係(波動関数がアッ
プ用、ダウン用になるので、自ずと電荷密度もアップ用、ダウン用が必要とな
ります。当然全電荷密度用の配列も必要です)、サブルーチンCONV2での電荷
密度混合用の作業配列変数、固有値(EKO)、バンドの占有数(OCCUP)、サブ
ルーチンFORCEなどで使用している作業用変数(ZFBB,ZFC)、交換相関項など
の配列(ここが一番面倒)がスピン用に書き換えたり、新たに作る必要があり
ます。
(7/7、1998)磁性を扱う場合、最初のアップ、ダウン別の初期電荷
密度は、どちらかが多くなるように設定します。この時、アップ、ダウンを同
じにするとパラマグネティック(非磁性)の計算と同等になります。もし、L
SDA計算が(LSD近似の範囲内で)正しく進むなら、最終的にアップ、ダ
ウン異なった電荷量になって収束します(分極化した方が安定なら)。磁性を
扱う場合のアップ、ダウンそれぞれの電荷密度の設定はアップ側を全体の75
%、ダウン側を25%にして計算しています(強磁性NI,Co,Feの場合)。この
設定はこれがベストという訳ではなく、50%、50%以外ならどのような設
定も可能です(が、あまり極端なものでは、計算が破綻したり、収束しない可
能性があります)。
(11/30、2004)反強磁性やフェリ磁性を扱う場合は、初期電荷の
設定にもう一工夫が必要にあります。それは反強磁性などに向かうように最初
から電荷密度の分布を反強磁性的なものにして置きます。実際にはこれによっ
て得られた反強磁性状態と他の磁気状態との全エネルギーの比較で、いずれの
磁気構造が安定か判定することとなります。初期値の設定によっては、反強磁
性的な分布を出発点としても、常磁性に落ち着いてしまう可能性もあります。
また、反強磁性を扱う場合その対称性が常磁性、強磁性と異なる(単位胞も2
倍以上となる)ものとなります。
(12/8、1999)磁気モーメントの求め方は、用語集をご参照下さい。
(7/7、2011)磁気構造の模式的な表現
参考図1
〔png形式、6 kb〕:摸式的な状態密度で、強磁性、常磁性、反強磁
性、ハーフメタリックを表現したもの。強磁性、常磁性、反強磁性の各状態は
必ずしも金属的である必要はない(=ギャップが空いてもかまわない)。注:
図とウェブページ上の文章とは必ずしも整合していない。
参考図2
〔png形式、7 kb〕:↑(Up spin)、↓(Down spin)で、強磁性、フェ
リ磁性、反強磁性、ノンコリニアな場合の各磁気構造を2次元的な平面上で表
現したもの(あくまで摸式的な表現であり、厳密性に欠けることに注意)。平
面を区切っている四角が最小の単位(単位胞)に相当。注:図とウェブページ
上の文章とは必ずしも整合していない。
(12/2、2004)スピン分極する場合の交換エネルギー部分について
以下に説明を行なう。
交換エネルギーEx(ρ)について、これは、
Ex(ρ) = ∫ρεx(ρ)dr ・・・(1)
と表現でき、εx(ρ)を交換エネルギー密度と言い、
εx(ρ) = Aρ1/3 ・・・(2)
とする。Aは適当な係数。(*)一様な電子ガスでの交換エネルギーの形は、
上記のようなものとなる。
従って、Ex(ρ)は、
Ex(ρ) = A∫ρ4/3dr ・・・(3)
となる。これをスピン分極(偏極)していない場合の交換エネルギーの表式
とする。次に、スピン分極した場合を考える。この場合の交換エネルギーを、
Ex(ρ↑,ρ↓)とすると、
Ex(ρ↑,ρ↓) = ○A∫{ ρ↑4/3 }dr + ○A∫{ ρ↓4/3 }dr
= ○A∫{ ρ↑4/3 + ρ↓4/3 }dr ・・・(4)
と表現される。○は未知の係数とする。常磁性状態の場合(ρ↑ = ρ↓ =
ρ/2)を上記分極した交換エネルギーの式に適用すると、
Ex(ρ/2,ρ/2) = ○A∫{ (ρ/2)4/3 + (ρ/2)4/3 }dr
= ○A∫{ 2(1/2)4/3ρ4/3 }dr
= ○2(1/2)4/3A∫ρ4/3 dr ・・・(5)
となる。この(5)の表式は、(3)の表式と一致するはずで、そのためには未知
の係数○が、○ = 21/3でないとならない。従って、スピン分極を
考慮した交換エネルギーの表式(4)は、
Ex(ρ↑,ρ↓) = 21/3A∫{ ρ↑4/3 + ρ↓4/3 }dr ・・・(6)
となる。ここで重要なのは、分極を考慮しない場合と考慮する場合で表式が
異なることである。従って、分極を考慮しない表式をそのままスピン分極する
場合に適用することは磁性状態に関して間違った結果を導く可能性がある。相
関エネルギー部分は表式がより複雑であり、分極の考慮、非考慮の使い分けは
より重要となる。
式(6)は、分極(偏極)度ζを使って、ρ↑=(1+ζ)/2, ρ↓=(1-ζ)/2から、
Ex(ρ↑,ρ↓) = (1/2)A∫ ρ4/3[ (1 + ζ)4/3 + (1 - ζ)4/3]dr ・・・(7)
とも書き表せる。ζ=0なら(3)式に帰着し、ζ=1(またはζ=-1)なら、(6)
式となる。
- 【参考文献】:磁性関連
- 「密度汎関数法とその応用‐分子・クラスターの電子状態」、菅野 暁監
修、里子允敏、大西楢平著、講談社サイエンティフィックの2.3節、4.5.
1節での説明。特に後者は交換相関エネルギーのスピン分極の表式がいくつか
掲載されている。
- 「原子・分子の密度汎関数法」、R.G.パール、W.ヤング著、狩野
覚、関 元、吉田元二 監訳、シュプリンガー・フェアラーク東京の8.2節及
び付録E。
- 「固体電子構造‐物質設計の基礎‐」、藤原毅夫著、朝倉書店の3.1.
2節に、von-BarthとHedin(1972年)によるスピン分極に対応した交換相
関ポテンシャルの表式あり。
- 「コンピューターでみる固体の中の電子」、和光信也著、講談社サイエ
ンティフィックの4.4節の46頁に、GunnarsonとLundqvist(1976)によるス
ピン分極に対応した交換相関ポテンシャルの表式あり。
- 上記論文に関しては、バンド計算関連重要論文ページも参照のこと。
- 「金属間化合物の電子構造と磁性 - 3d-pnictidesを中心として
- 」、望月和子・井門秀秋・伊藤忠栄・森藤正人著、大学教育出版
(2/17、2005)電子ガスにおける交換エネルギー、交換ポテンシャ
ル部分の係数に関して以下に説明を行なう。この時、スピンは考えないことに
する。単位は、ハートレー原子単位を
用いる。従って、e2 = 1である。交換エネルギーEx
(ρ)について、これは(1)式より、
Ex(ρ) = ∫ρεx(ρ)dr ・・・(1)
と表現でき、交換エネルギー密度εx(ρ)は、
εx(ρ) = Aρ1/3 ・・・(2)
ということは既に(2)式で示した。Aは適当な係数であり、局所密度近似下に
おける一様な電子ガスでの係数Aは、
A = -3/4 (3/π)1/3 ・・・(8)
である。この場合、Aの値(絶対値)はおおよそ、0.7386...となる。上式の
導出に関しては、バンド計算関連の参考文献、
書籍等を参考にして欲しい。交換エネルギー密度εxは、rsパ
ラメータ、フェルミ波数それぞれで表現すると、
εx = -3/4 (3/π)1/3 ρ1/3 ・・・(9a)
= - 0.4582 / rs ・・・(9a)
= - (3/4) kF ・・・(9c)
となる。この時、
rs = (3/4)1/3 (1/π)1/3 (1/ρ)1/3 ・・・(10)
kF = (3 π2 ρ)1/3 ・・・(11)
である。交換ポテンシャルは、
Vx(r) = d(ρεx(ρ))/dρ ・・・(12)
から求められるので、
Vx = d(ρεx(ρ))/dρ = - d( (3/4)(3/π)1/3 ρ4/3)/dρ
= - (3/4)(4/3)(3/π)1/3 ρ1/3
= - (3/π)1/3 ρ1/3(r) ・・・(13)
となる。この時、ρ1/3に付く係数(絶対値)(3/π)
1/3はおおよそ、0.98475...(先のA値の4/3倍)となる。以上は、
ハートレー原子単位で考えている。上
記の式には表示されていないが、ハートレー原子単位では、e2 =
1であるが、リュードベリ単位では、e2 = 2である。また、スレー
ターによる交換ポテンシャルは上記交換ポテンシャルの3/2倍となる。Xα近似
では係数αがつき(Vxαとする)、
Vxα = αVx,sl = (3/2)αVx ・・・(14)
となる。Vx,slはスレーターの交換ポテンシャル(= 3/2 Vx)、αは通常、
(2/3)〜1までの範囲をとる。α = 2/3がVxであり、α = 1でVx,slとなる。
スピン偏極を考える場合は、交換エネルギーεxに21/3が付く。
ほとんどの式で省略しているが、Vx = Vx(ρ) =Vx(ρ(r)) = Vx(r)、εx = ε
x(ρ) = εx(ρ(r)) = εx(r)、ρ = ρ(r)である。
尚、以上の議論は、分子のおも
ちゃ箱の掲示板での議論が元となっている(←係数の問題は重要であり、
覚書として記す必要、意義があると考えた)。
(3/1、2005)電子ガスにおける交換相関エネルギーExc(交換相関
項、交換相関エネルギー項とも言う)を、
Exc[ρ] = ∫ρεxc(ρ)dr ・・・(15)
とするのが局所密度近似である。ρ
は電荷密度(ρ = ρ(r)、rは座標で、r = r)である。上式左辺の括
弧が[]となっていて、密度汎関数法に
おいて厳密に求められるべきものであるが、右辺側は近似的に、ρεxc(ρ)と
いうように局所的なρ(r)に関しての体積積分で求まるとしている(左辺→右
辺における、[]→()は重要)。勿論、厳密なExc[ρ]を現時点で求めることは
不可能である。求める式も方法も手段も全く未解決である。局所密度近似は、
上式右辺のようにして近似的にExcを求めるものである。交換相関エネルギー
密度εxc(電子1個当たりの交換相関エネルギーと表現する場合もある)の表
式も、近似的に求められたものが使用されている。電子ガスでの低密度、高密
度からの内挿(または外挿)による方法や、RPAを用いて求めたもの、モンテ
カルロ法から求めたものなど、εxcにはいくつかの表式が存在する。スピンを考慮しない範囲で
は、それぞれの表式で大きな差はみられないが、場合によりあまり良い結果を
与えないこともある。また、磁性が絡む場合
も注意が必要(調査中)。
交換相関エネルギー密度εxc(ρ)が電子1個分に相当すること
の(あまり厳密でないかもしれない)説明。電荷密度ρが一様であるとすると、
ρ(r) = ρ = N/Ω ・・・(16)
となる。Nは系(単位胞内)の全電子数。Ωは系(単位胞)の体積である。
こうすると、(15)式右辺は、
∫ρεxc(ρ)dr = ∫(N/Ω)εxc(N/Ω)dr
= (N/Ω)εxc(N/Ω)∫dr = (N/Ω)εxc(N/Ω)Ω = Nεxc(N/Ω) ・・・(17)
となる。以上から、
εxc(N/Ω) = Exc/N ・・・(18)
であり、交換相関エネルギー密度は電子1個分の交換相関エネルギーとなっ
ている。以上は、ρが一様(一定)であることを前提としているが、実際の局
所密度近似も電荷密度の空間的な変化が緩やかであることを前提とした近似で
ある。空間上で激しく電荷密度が変化するとしたら、局所的な記述が破綻する
ことは容易に想像できる。
局所密度近似では不完全な形で相関項を取り入れるため、ハートリー・フォッ
ク近似の段階では厳密に相殺される、電子自身が自分と相互作用する自己相互作用部分が残ってしまう。このため無限遠
で感じる電子のポテンシャル(一電子ポテンシャル)が、-1/rにならずに指数
関数的に減衰してしまう。関連用語:自己
相互作用補正(SIC)
(相関項の由来)
系の全エネルギーEtotが厳密に得られているとして、ハートリー・フォック
近似で得られる当該する系の全エネルギーを、EtotHFとする。相
関項Ecは、
Ec = Etot - EtotHF ・・・(19)
として表現される。つまり厳密な全エネルギーの値とハートリー・フォック
近似によるものとの差の部分が相関項(相関エネルギー)となる。相関項は勿
論、(クーロン項+交換項以外の)電子間の相互作用を記述する項である。
(関連)相互作用していない系での運動エネルギー項←相互作用する部分は、
全て相関項に押し込める。相互作用していない電子の運動エネルギーなので、
ビリアル定理での扱いにも気を付ける必要
がある(調査中)。
(注意)相関項の考え方は、必ずしも上
記の定義の仕方に縛られない(上記以外の説明、定義のされ方がなされる場合
がある。調査中)。
(局所とは)
Exc = ∫ρ(r)εxc(ρ(r))drと、局所的な座標r(= r)のみに依る形
の状態を、”局所”と言う(数学的、物理的には不確かな説明かもしれない)。
もし、Excが、
Exc = ∫f(r,r')drdr' ・・・(20)
のような形で表現される場合、最早座標rのみによる局所的な記述は出来な
くなる。これは”非局所”なものとなっている。つまりその場(の座標
r)だけでExを決めることは出来なくなっている。局所、非局所と言う
用語は上記と異なる意味合いで使われることがある(調査中)。関連用語:局
所擬ポテンシャル、非局所擬ポテンシャル
(密度汎関数法〔または密度汎関数理論〕は変分問題)
変分パラメーターは(電子の)電荷密度。密度汎関数法は、絶対零度での変
分問題。従って全エネルギー(=内部エネルギー)の電荷密度に関しての変分
となる(Kohn, Shamの論文)。有限温度
への拡張は、Merminによるものが最初(論文
)である。この場合、全エネルギーでなく自由エネルギーに対する変分と
なる(変分パラメーターは電荷密度)。ただ、バンド計算で具体的にどのよう
な処方を用いればよいかに関しての言及はされていない。実際に有限温度をバ
ンド計算で扱うのは、大変難しい。一つの処方としてはバンドにフェルミ分布
関数による広がり(ブローデニングと同じ方法)を持たせて、その広がりに関
するパラメーターを変分の対象として扱うことが行なわれている(有限温度LDA、調査中)。
関連ページ:[密度汎関数法、局所密度近似関連
情報]
(4/8、2005)これに関しては、Lecture Notes in Physics, A
Primer in Density Functional Theoryの5. R. W. Godby and
P. Garcia-Gonzalez, "Density Functional Theories and Self-energy
Approach"の196〜199頁(5.3.1節)の部分、福本先生に「よるレビュー(豊田
中央研究所 R & Dレビュー、Vol. 25, No. 4)、浜田先生、大西先生による”
日本物理学会誌”での解説(「固体の電子状態」、第40巻、第11号(19
85)、842頁の部分)、寺倉先生、浜田先生による雑誌”固体物理”での
解説(「バンド計算法の最近の発展(II) - 密度汎関数法 -」、第20巻、
第9号(1985)、706頁の部分〔第5節〕)、里子先生等の著書(「密度汎関数法とその応用‐分子・ク
ラスターの電子状態」)などに関連する記述がある。更に参考文献として、
[1] J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz, Jr., Phys. Rev. Lett., Vol. 49, No. 23, 1691(1982)
[2] L. J. Sham amd M. Schlüter, Phys. Rev. Lett., Vol. 51, No. 20, 1888(1983)
などが挙げられる(福本先生のレビューでの参考文献も有用)。
以下、スピンは考えず、バンドギャップの存在する半導体、絶縁体を対象と
する。スピンを考えないので、一つの準位(または軌道、バンドと言ってもよいだろう)に収容でき
る電子の数は2である。まず、電子親和力をA、及びイオン化エネルギー(イ
オン化ポテンシャル)をIとすると、
A = Etot,N - Etot,N+1 ・・・(21)
I = Etot,N-1 - Etot,N ・・・(22)
である。Etot,NはN個の電子からなる系の基底状態での全エネル
ギーを意味する。上記の電子親和力とイオン化エネルギーから、バンドギャッ
プEgapは、
Egap = I - A ・・・(23)
と表される。このEgapが、実際の実験等で観測されるバンドギャッ
プに相当する。但し、実験手法などによりバンドギャップの解釈は微妙に異な
る場合がある。実験が一体何を観測しているのか、そしてその時のバンドギャッ
プの定義なり解釈なりの議論は大変重要であるが、ここではその詳細を述べる
だけの力量が筆者にないため、これ以上の言及は避けることとする。
一方、このEgapをKohn-Shamの密度汎関数法で表現すると、
Egap = εKS,N+1(N+1) - εKS,N(N) ・・・(24)
となる。上の式で、εKS,N+1(N+1) は、N+1電子系でのN+1個目
の準位(軌道、バンド)、つまり最高占有準位(バンド、軌道またはコー
ン・シャム軌道)である。εKS,N(N)は、N電子系でのN個目の
準位(軌道、バンド)である。これはN電子系での最高占有準位(軌道、バン
ド)になっている。そして、実際のコーン・シャム形式での密度汎関数法に基
づくバンド計算で得られるバンドギャップは、
εKS,gap = εKS,N(N+1) - εKS,N(N) ・・・(25)
となる。εKS,N(N+1)は、N電子系でのN+1個目の準位(軌道、バ
ンド)、つまり空の準位(軌道、バンド)である。以上から、
EgapとεKS,gapとの関係が導かれ、それは、
Egap = (εKS,N(N+1) - εKS,N(N)) + (εKS,N+1(N+1) - εKS,N(N+1))
= εKS,gap + Δxc ・・・(26)
となる。ここで出てくるΔxcは、
Δxc = εKS,N+1(N+1) - εKS,N(N+1) ・・・(27)
であり、これはN+1電子系でのN+1個目の最高占有準位(軌道、バンド)と、
N電子系でのN+1個目の空(非占有)の準位(軌道、バンド)との差となってい
る。N電子系に対してバンド計算を行なった場合、電荷中性の要請からそもそ
も、N電子系に1個だけ電子を加える(=N+1個系)ような計算が出来ない。周
期的境界条件下での単位胞(またはスーパーセル)で、電子を1個だけ加えれ
ば、エバルト項などが発散する。これの対処としてのジェリウムモデルの導入は、元のN電子系との整合性
が崩れ、上記(式26など)にある全エネルギー同士の比較が意味をなさなくな
る。
Δxcの起源に関しては、これをN-δからN+δ(Nは整数:電子の
数、δは微小量〔=無限小〕)における交換相関ポテンシャル部分の不連続な
とびによるとする説明が、先の文献[1][2]等でなされている。つまり、
Δxc = vxc(N+δ) - vxc(N-δ) ・・・(28)
である。vxcは、交換相関ポテンシャル。また、δExc/δρ=vxcより、
Δxc = δExc[ρ]/δρ|N+1 - δExc[ρ]/δρ|N + O(1/N) ・・・(29)
となる。ここでNは十分大きな量と考えている(N ≫ 0)。Nが十分大きけれ
ば、誤差の部分Oは無視できることとなる。式28にも本当は誤差部分
が存在する。式28→式29に関しては、筆者自身まだ十分理解できていない
部分あり(←調査、勉強中→)。Nが十分大きい(=無限大)ということと、
δが無限小であるということは、いずれもN+1(N → ∞)、N+δ(δ → 1/∞)
での軌道緩和効果が事実上無視できることを意味している。
Δxcが正の有限な量であれば、通常のバンド計算によるバンド
ギャップは常に過小評価していることが示せる。交換相関ポテンシャルの非連
続的なとびによる過小評価が、全体としてのバンドギャップのバンド計算によ
る誤差の8割方を占めるということである。残り2割の中には、軌道緩和の効
果などが含まれるものと考えられる。系が金属だった場合、Δxc
は、ゼロか負の値である必要がある(正ならギャップが生じ得るし、負の値だっ
たとしてもそう大きな値にはなり得ない)。但し、Δxc > 0は、
今の段階で筆者にとっては自明ではない(←調査、勉強中)。更に、第一原理
的かつ簡便にΔxcを定量的に評価することは困難である。これを
解決するには、局所密度近似を越える試み
における自己相互作用補正や、GW近似などを考える必要がある。
【おまけ】ヤナックの定理より、i番目
のコーン・シャム軌道をεiとし、その占有数をfi(0
< fi < 1とする)となる。この時、イオン化エネルギーIは、
I = Etot,N-1|fi = 0 - Etot,N|fi = 1 = - ∫0 → 1 εi(f) df ・・・(30)
となる。積分部分、∫0 → 1 εi(f) dfにおいて、
∫0 → 1 εi(f) df ≒ εi(0.5) ・・・(31)
とし、I = -εi(0.5)とするのがSlaterの遷移状態理論(近似)
である。31式導出に関しては小口先生の著書「バンド計算」(内田老鶴圃刊)
3.4節参照。"0.5"は占有数が半分であることを意味する。ここで、一つの
KS軌道(準位、バンド)の収容電子数(1か2)のことは深く考えないことに
する。
現在(6/1、1999)、Perdew先生、Burke先生、Ernzerhof先生による
PBEコードを入手、revpe_d.fに導入する作業を遂行中です。
PBEに関しての[参考文献]、Perdew先
生の[ウェブページ](PBEコード入
手に関して深く感謝する次第です)。
(7/16、2001)現在のPBEコードのある[ところ](Rutgers大学のKieron Burke
先生のサイト、”past projects”のページにPBEコードへのリンクがある。使
用方法、諸注意は当該ページ、データ等のドキュメント参照のこと)。
GGA導入に関して、現在プログラムへの導入作業はほぼ終了し、バルク系、
表面系に対するテストを行なっています。これらの顛末については、現在執筆
準備中(*)であります。
(2/1、2000)現在、仮想的なBCC構造(立方体のユニットセル内
に2個原子が存在)でのAlのテスト計算を行ない、原子位置を平衡位置から
少しだけずらせる(いくつかの場合で検証)ことによって生じる力について
[検証]しました。
検証の結果、力は(一部の場合、k点数を十分にとる必要あり)十分な精度
で正しく得られていることを確認しました。この場合、力は線形近似(調和近
似つまり”ばね”)の範囲内で十分記述され、全エネルギーの位置変位による
変化分=1/2×F×Δxとなります(F:力、Δx変位長さ)。F=kΔxな
ので変位の長さを1/10にするとエネルギーの変化量は1/100になりま
す。
少し状況報告(6/25、1999)。GGAでは電荷密度に対して微分を
行なうため、特に表面系のようなスーパーセル+スラブ近似を行なっている場
合、真空層での電荷密度がほとんどゼロかそれに近い部分での微分対しての寄
与について考えなければなりません。バルク系の計算では、さして計算に問題
は生じなかったのですが、表面系では、全エネルギーも原子間に働く力も、バ
ルク系で扱った手順、条件と同じではまともに計算できないことが判明しまし
た。
(2/19、2003)現在も少しずつバグ等を潰しつつ作業はゆっくりと
進行中。最近、グラファイトの格子定数をGGAではまともに求められないと
いう話を聞いて、こちらでも試してみる(GGAの関数形は、PBE)。結果
としてGGAでは、c軸方向の格子定数が求められなかった(バルクでいるよ
り、単独のグラファイトシート〔単層膜〕の方がより安定となってしまう)。
LDAでは、格子定数は求まるが、採用する関数形によって合う場合と、全
く合わない(Wigner形式では、c軸の格子定数が数%以上伸び目に出る)場合
があり、また精度を上げよう(エネルギーカットオフを上げる)とすると、む
しろ結果が悪くなってしまう(筆者の計算での話)。
GGAでは、van der Waals結合を正
確に表現することが特に困難で(勿論、LDAでも困難)、他に、窒素の分子
性結晶、グラファイトの面間距離(c軸の格子定数)など、van der Waals力に
よって結合している物質の格子定数を正確に求められないことが分かっている。
グラファイト類似の面間の結合が弱いと推察される系(炭素-ホウ素化合物)
でも、やはりGGAで平衡面間隔が求められない事例あり。局所密度近似では
最小値、つまり平衡面間隔が存在する。GGAでは面間隔が20 a.u.以上とい
う値にしてもどんどん全エネルギーが下がっていく。→つまり、バルクでなく
単独の一平面である方がより安定と結論される(7/15、2004)。
(11/5、2003)GGA計算に関しては若干の実践的進展あり。HBC
(LiBC構造と同じ結晶構造、詳細はLiBC[ページ
]参照)に関して、PBE形式でのGGA計算結果をIUMRS-ICAM2003にて発表。
まだ、GGAの基での圧力の導出は出来ていないので、格子定数を少しずつ変
えながら全エネルギーの最小値を探索するという原始的手段を用いている。結
果は、それなりに正しい模様、詳細はIUMRS-ICAM2003のプロシーディングス発
刊(K. Kobayashi, M. Arai and T. Sasaki, "Lattice Anomalies of
MBC (M = H, Li, Na) Under Anisotropic Compression",
Trans. MRS-J, Vol. 29, No. 8, 3799 - 3802 (2004)[IUMRS-ICAM2003])以降
に公開。
以上(GGA導入)に関しての対処方法について今後も述べる予定(現在も
なお調査、検討中、7/16、2000→2/19、2003→11/5、2
003→7/15、2004)。
【参考文献】
[1][PBE]J. P. Perdew, K. Burke and M. Ernzerhof:
Phys. Rev. Lett. 77 (1996) 3865[PBE].
[2]J. P. Perdew and W. Yue, Phys. Rev. B33, 8800(1986)[(24)式:
GGA exchange potential]
[3]J. P. Perdew, M. Ernzerhof, A. Zupan and K. Burke,
J. Chem. Phys. 108, 1522(1998)[Appendix A: GGA correlation
potential]
[4]J. A. White and D. M. Bird, Phys. Rev. B50,
4954(1994)[Implementation of gradient-corrected exchange-correlation
potentials]
[GGAにおけるストレス関連]
[1]A. D. Corso and R. Resta, Phys. Rev. B50, 4327(1994)
[2]L. C. Balbas, J. L. Martins and J. M. Soler, Phys. Rev. B64,
165110(2001)
[3]N. Nagasako and T. Oguchi, Journal of the Physical Society of
Japan, Vol. 82, No. 4, 044701(2013)[Stress formulation][All-electron
FLAPW][Accuracy check][GGA]
[4]L. C. Balbas, J. L. Martins and J. M. Soler, Phys. Rev. B64,
165110(2001)
[5]N. A. W. Holzwarth先生(Department of Physics, Wake Forest
University)のところに
ある、"Notes on GGA implementation"(PDFによるノート)は大変有用。
一般化された密度勾配近似という名称からも分かるように、GGAでは電荷
密度ρ(r)の微分(▽ρやΔρなど)が必要となります。数値微分を使って電
荷密度の勾配を求めることも可能ですが、GGAでは普通フーリエ変換(実際
には高速フーリエ変換〔FFT〕)を使用して勾配を求めます。これは通常、数
値微分よりフーリエ変換を利用する手法の方が、精度的、速度的に有効なため
です。この手法は、実空間での電荷密度ρ(r)の逆フーリエ変換(←以下の式
〔G → r〕を逆変換と定義)は、
ρ(r) = ΣGρ(G)exp(iGr)
となります。G:逆格子ベクトル、r:実格子ベクトル、i:虚
数。ρ(G):実空間での電荷密度をフーリエ変換した(逆格子空間上で
の)電荷密度。これに対する勾配の絶対値|▽ρ(r)|は、
|▽ρ(r)| = |iΣGGρ(G)exp(iGr)|
となります。上式でのexpの肩の部分でrに関して微分します。
rに依るのはこの肩の部分のみです。つまり、iGρ(G)
に対する逆フーリエ変換をすれば、|▽ρ(r)|を求めることが出来ます。同
じ要領で、Δρ( = ▽2ρ(r))なども求めることができます。
希土類金属擬ポテンシャルにアプローチするためには、スピン軌道相互作用
を考慮しなければならないということで、摂動形式で導入することを計画しま
したが、希土類金属くらい重い原子での計算ではスピン軌道相互作用は摂動的
な取り扱いでは無理ということが判明(これは、金材研での鈴木修吾先生のセ
ミナーで強く認識しました)しました。従って、摂動形式での導入だけでは駄
目であることが分かり、現在作業、計画共頓座しています(6/1、1999)。
比較的軽い原子である、In、Sb、Snなどでは、スピン軌道相互作用によって
バンド構造におけるバンドの分裂が起こりますが、格子定数や体積弾性率など
は、スピン軌道相互作用による影響をほとんど受けません。実際、我々の扱っ
た、InSb/Sn界面の計算(神奈川工
科大学の山本氏との共同研究)ではスピン軌道相互作用は考慮していませんが、
格子定数、体積弾性率は実験値と十分一致(InはPCCを考慮しても尚2%以上の格子定数
のずれがありましたが、InSbとしてなら、実験値と良く一致)する結果が得ら
れています。
このようなIn、Sb、Sn辺りでは摂動でスピン軌道相互作用を導入することに
より、より精度の高いバンド構造を得ることが可能と思われます。またこれに
よる格子パラメーターへの寄与はほとんどないと思われます。これだけで断定
はできませんが、構造最適化においても、スピン軌道相互作用は(バンド分裂
の影響が出てくるような場合を除いて)ほとんど影響しないだろうと思われま
す(確証なし、調査中)。
ただ、他にも幾つか問題があり、実際にバンド計算する段階まで全く達して
いません。特に、スピン軌道相互作用用擬ポテンシャルを作成する目処が全く
立っていません(これでは計算にとりかかれるはずもない)。
擬ポテンシャル+平面波によるスピン軌道相互作用の摂動による表式は、次
節で示してある参考文献から得ることが可能ですが、これをさらに具体的に表
したものを示します(png画像、30KBほど
です。方向を修正し見易くしました。6/21、1999)。
(注意)この表式は、計算による確認等を全く行なって
いません。従って、本当に正しいかどうかは不明です(筆者及び物質・材料研
究機構は一切責任を負えません)。
筆者が現在、把握しているものを以下に示します(当然、全てを網羅してい
る訳ではありません。擬ポテンシャルが関連しているものが中心です)。
- P. J. Lin and L. Kleinman, Phys. Rev., Vol. 142, No. 2, 478(1966)
- G. Weisz, Phys. Rev., Vol. 149, No. 2, 504(1966)
- L. R. Saravia and D. Brust, Phys. Rev., Vol. 176, No. 3,
915(1968)
- J. R. Chelikowsky and M. L. Cohen, Phys. Rev. B14,
556(1976)
- M. S. Hybertsen and S. G. Louie, Phys. Rev. B34,
2920(1986)
- M. P. Surh, Ming-Fu Li and S. G. Louie, Phys. Rev. B43,
4286(1991)
- L. A. Hemstreet, C. Y. Fong and J. S. Nelson,
Phys. Rev. B47, 4238(1993)
- T. B. Boykin, Phys. Rev. B57, 1620(1998)[TB]
- G. Theurich and N. A. Hill, Phys. Rev. B64,
073106(2001)[Self-consistent treatment][Relativistic][Fully separable
PS]
- M. D. Stiles, S. V. Halilov, R. A. Hyman and A. Zangwill,
Phys. Rev. B64, 104430(2001)[Spin-other-orbit
interaction][Magnetcrystalline anisotropy]
- X. Gonze, et al, Computational Materials Science 25, 478(2002)の
2.4 Spin-orbit couplingの節
- A. D. Corso and A. M. Conte, Phys. Rev. B71,
115106(2005)[UPS][Au][Pt]
- E. Fromager, L. Visscher, L. Maron and C. Teichteil,
J. Chem. Phys., Vol. 123, No. 16, 164105(2005)[One-component PS
SOC][Accuracy][CI]
- T. Oda and A. Hosokawa, Phys. Rev. B72,
224428(2005)[Fully relativistic two-component-spinor][SOI][UPS][PW]
- Jacques K. Desmarais, Alberto Boccuni, Jean-Pierre Flament, Bernard Kirtman, Alessandro Erba, arXiv:2302.06143[Perturbation theory treatment][Spin-orbit coupling][Coupled perturbed method][Solid]
- 柳瀬陽一、播磨尚朝、初等固体物理講座:”スピン軌道相互作用と結晶
中の電子状態(その1)孤立原子におけるスピン軌道相互作用の定量的評価”、
固体物理、第46巻、第5号、229(2011)
”(その2)空間反転対称性が破れた系の反対称スピン軌道相互作用”、固
体物理、第46巻、第6号、283頁(2011)
”(その3)”空間反転対称性が破れた系の反対称スピン軌道相互作用(応
用編)”、第47巻、第3号、101頁(2012)
- 播磨尚朝、「結晶内電子状態のスピン自由度による縮退と分裂」、日本物理学会誌、第78巻、第5号、254頁(2023)
- 新田淳作、「スピン軌道相互作用とスピントロニクス」、応用物理、第92巻、第6号、324頁(2023)
以上の他に、(旧)電総研(産総研)の織田先生の博士論文(高圧化でのヨ
ウ素の計算で、スピン軌道相互作用を考慮した計算も行なっている)も参考に
なりました。擬ポテンシャルの計算は摂動によるものです。
[先頭][総目次
][最初に戻る][使ってみよう編][バンド屋さんマップ][密度汎関数法、局所密度近似関連情報]
{kind=link}