[遷移金属炭化物、窒化物表面関連情報]
(Start, 2/5, 2002)(Update, 4/30, 2021)[Top]
計算対象:遷移金属炭化物、窒化物表面(001)面
岩塩構造(NaCl型構造)を持つ、TiC、ZrC、NbC、HfC、TaC、TiN、ZrN、NbN、HfN、TaNの各表面で、(001)面の構造最適化とその電子状態を求める計算を行なった。計算方法は、第一原理分子動力学(FPMD)手法。
(001)面は、無極性な面となっており、陽イオン(遷移金属原子)と、陰イオン(炭素または窒素原子)が、それぞれ同じ数だけ交互に配置している。実際の計算の上でも極性面〔(111)面など〕を扱うより、無極性面の方が計算が楽である。これは無極性面上では電場勾配が生じないためである(通常のバンド計算では周期的境界条件のため、電場を扱うのが大変難しい)。
表面は、スラブ近似によって周期的境界条件の下で扱う。バンド計算手法を使って仕事関数を求める方法については、用語集の”仕事関数の計算”を参照。
筆者(小林)の研究
(ランプリング [rumpling])
この岩塩構造物質の(001)表面は無極性面となっていて、ランプリングという構造変位を起こす。おおまかに言えば、表面緩和の過程で陽イオンより陰イオンの方が大きい分、バルク側への変位が陽イオンより小さくなる。従って、陽イオンはバルク側、陰イオンは(相対的或いは絶対的に)真空側へ変位することとなる(必ずしもそうならない場合もある)。
遷移金属炭化物[1]、窒化物[2]において、筆者が計算した系では全て炭素、窒素原子(陰イオンに相当)が真空側、遷移金属原子(陽イオンに相当)がバルク側に変位した。電子状態はバルク、表面(理想表面構造、最適化構造共に)とも金属的であった。
構造最適化以外に仕事関数を理論計算の結果から求めた。計算の結果、遷移金属炭化物表面より窒化物表面の方が、仕事関数の値が小さくなることが分かっている[2]。その理由に関しては、参考文献[2]において説明されているが、非常に手短に言えば炭素と窒素の価電子数の差によるものと考えられる。更に、遷移金属窒化物表面では、理想表面より構造最適化した表面(←ランプリングにより窒素が真空側に変位している)の方が、仕事関数の値が小さく(低く)なる場合があることが分かった。一方、筆者が計算した炭化物表面では、全ての場合で理想表面の方が小さな値となる。計算で得られる仕事関数の精度は、文献[1][2](特に文献[2])にあるように、実験値との一致は比較的良いと言える。実験との差は大体、0.2〜0.3 eV程度であり差が最も大きいのはHfN(001)表面で、約1 eVの差がある。
(MgO基盤表面上のTiNドット構造)
TiN/MgO-1x1界面構造を最初に扱った[4][5][6]。更に、より大きなMgO基盤上に、TiNの板状及び箱状ドット構造を乗せた場合を扱った[7][8][9]。2019年3月時点での最大のMgO基盤サイズは、6x6である(文献では5x5まで)。TiN/MgO-1x1界面構造では、系の電子状態がTiNの層数に関らず金属的となった[5]が、MgO-2x2より大きなサイズの基盤上に、TiNドット構造を乗せた系は、(一部を場合を除いて)半導体的になることが分った[7][8][9]。電子状態は、MgO基盤のサイズ、TiNドットの形状に依存することも明らかとなった。ScN/MgO(ScNドット)超格子構造の計算も行った[10]。
特に、TiN dot/MgO超格子の電子状態(電子構造)に関して、(電子状態が半導体的な場合)状態密度において、ギャップ近傍にあるピークなどは、Tiの3d軌道からの寄与が主なものとなっている(詳細は、文献[9]参照)。
更に、ScN dot/MgO超格子の電子状態(電子構造)に関して、(電子状態が半導体的な場合)状態密度において、ギャップ近傍にあるピークなどは、TiNドットの場合と異り、Nの2pとScの3d軌道両方からの寄与が主なものとなっている(詳細は、文献[10]参照)。
(参考文献)
[1] K. Kobayashi, "First-principles study of the surface electronic structures of transition metal carbides", Jpn. J. Appl. Phys. Vol. 39,
No. 7B, (2000) 4311.
[2] K. Kobayashi, "First-Principles Study of the Electronic Properties of Transition Metal Nitride Surfaces", Surface Science 493 (2001) 665 [DOI: 10.1016/S0039-6028(01)01280-8](*).
[3] K. Kobayashi, "First-principles study of the TiX(X = B, C, O, N and F) surfaces", in proceedings of JK2000, (2001) 285. [PDF](PDF形式、174 kb)
[4]K. Kobayashi, N. Kobayashi, and K. Hirose, "First-Principles Study of TiN/MgO Interfaces", e-J. Surf. Sci. Nanotech., 12 (2014) 230 - 237 (ACSIN-12 & ICSPM21) [DOI: 10.1380/ejssnt.2014.230].←MgO(岩塩構造)の[バンド構造](png, 25.6 kb)
[5]K. Kobayashi, H. Takaki, N. Kobayashi, and K. Hirose, "Electronic Band Structure of Various TiN/MgO Superlattices", JPS Conf. Proc. 5 (2015) 011013 (CSW2014).
[6]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi and Kenji Hirose, "First-principles calculations of thermoelectric properties of TiN/MgO superlattices - the route for enhancement of thermoelectric effects in artificial nanostructures", J. Appl. Phys. 119 (2016) 014302.
[7]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic band structure of TiN/MgO nanostructures", Jpn. J. Appl. Phys. 56[4S] (2017) 04CK06 (Special issue).
[8]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Enhancement of thermoelectric properties in surface nanostructures", J. Electron. Mater. 46[10] (2017) 5593 - 5598(https page).
[9]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic Band Structure of TiN/MgO-4×4 and 5×5 Nanostructures", Jpn. J. Appl. Phys. 58[SB] (2019) SBBH06 (Special issue).
[10]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic and Lattice Properties of Nanostructured TiN/MgO and ScN/MgO Superlattices", Jpn. J. Appl. Phys. 60[SE] (2021) SE1006 (Special issue).
- 【バンド計算関連情報】
- TMC、TMN表面関連(バンド計算関連)[論文]
- 【関連】TMB2(遷移金属二ホウ化物)表面関連[論文]
- (第一原理)バンド計算における仕事関数の[計算の仕方]
- ↑関連:表面系のバンド構造にギャップがある場合の[一考察](まだ未完成+調査、勉強中)
- 仕事関数関連:双極子モーメントの[計算](+[関連ページ])
-
- (第一原理)バンド計算における極性のある表面の[扱い]
-
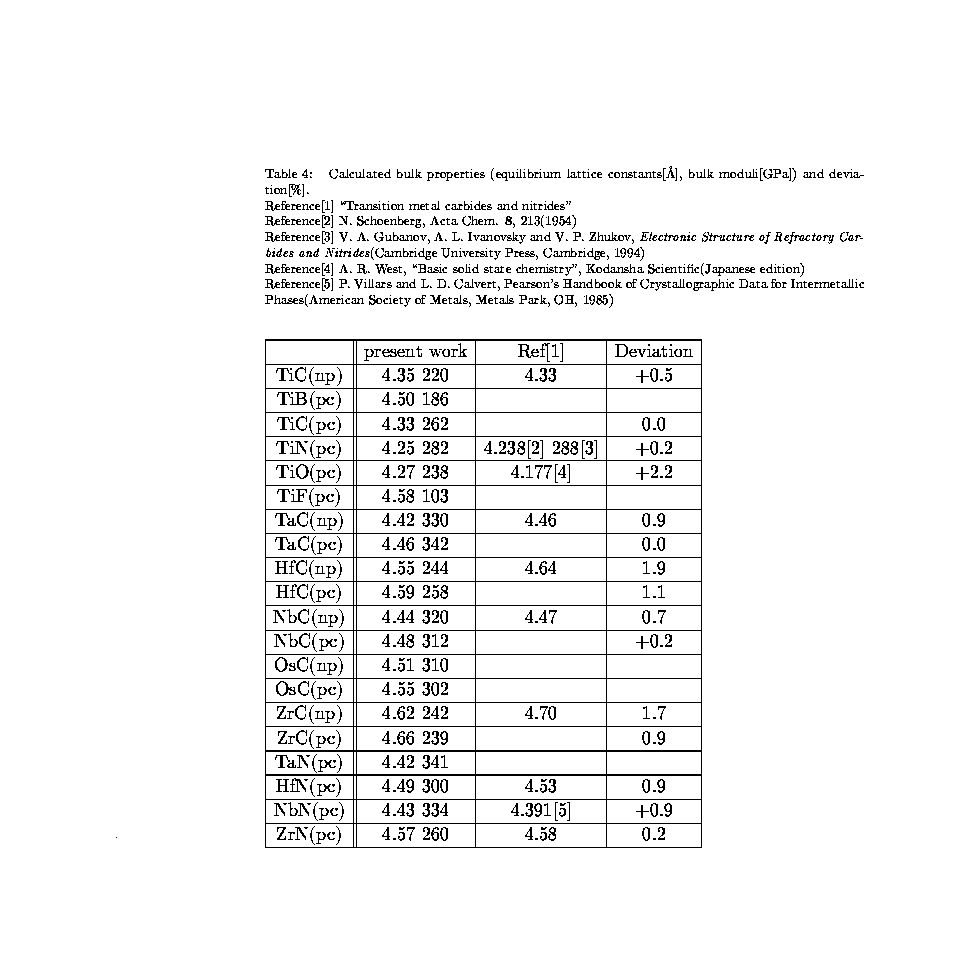
- 筆者が計算した遷移金属炭化物、窒化物(バルク)の平衡格子定数、体積弾性率の[データ]
-
- 遷移金属炭化物、窒化物の[バンド構造](バンド構造図集ページへ)
-
- 【表面関連情報】
- (実験関連情報)
- 有機EL材料の電子特性・光学特性の同時測定を大気中で実現:機構のプレスリリース(8/27、2012) ← バンドダイアグラム(真空準位を基準とした、VBM、CBMの位置関係)の観測が可能。
- (文献情報:教科書等)←筆者が所有するものが中心
- 塚田捷著、「表面物理入門」、東京大学出版会
- 塚田捷編、「表面における理論I - 構造と電子状態 -」、丸善株式会社(IIもあり)
- 村田好正著、「表面物理学」、朝倉書店
- R. Hoffmann, "Solids and Surfaces - A Chemist's View of Bonding in Extended Structures -"(邦題:固体と表面の理論化学)、小林宏、海津洋行、榎敏明共訳、丸善株式会社
-
- (文献情報:総説、解説論文等)
- 草部浩一、幾野佑一、長柄一誠、「密度汎関数法による仕事関数の決定に関して」連載企画(3) 〜第一原理計算(応用編)〜、表面科学、第29巻、第5号(2008年5月)、321 - 324頁
- 兵頭俊夫、長島泰之、「陽電子プローブ(I)白色陽電子の利用」、固体物理、第43巻、第2号、63 - 72(2008) ← 陽電子親和力、金属表面での(陽電子の)仕事関数に関する記述あり。
- 雑誌「表面科学」、第29巻、第2号(2008年2月)、特集:界面エレクトロニクス ← 仕事関数関連の記事あり(有用)。
- 吉武道子、「デバイス電極材料と仕事関数」、応用物理、第76巻、第4号(2007年4月)、399 - 404頁
- S. Yamamoto, "Fundamental physics of vacuum electron sources", Rep. Prog. Phys. 69 (2006) 181-232.
- M. Yoshitake, "Generic trend of work functions in transition-metal carbides and nitrides", J. Vac. Sci. Technol. A 32 (2014) 061403.
- 石田浩、「アルカリ金属と仕事関数」、固体物理<計算物理>特集号、第24巻、第3号、138 - 142(272 - 276)頁
-
- 筆者D論(PDF形式、320 kb、bandstructure.jpページへ):Si(001)表面に吸着したアルカリ金属(Na,K)の安定位置とその電子状態を計算した。第一原理分子動力学計算(方法論)やノルム保存型擬ポテンシャルについての記述もあり。
-
- (筆者の表面研究歴)
- 1990年前後:筆者D論の研究(↑上記参照↑)
- 1993年〜1994年:ダイヤモンド表面の計算
- 1999年〜現在:遷移金属炭化物、窒化物表面の計算(本ページ参照)
- 他に、山本氏(神奈川工科大学、筆者との共同研究)による計算(Sn/InSb界面の計算、TaB2、HfB2(0001)表面の計算等)がある。
-
- その他、バルク系の計算を主に行なった。例:[BC系][BN系(+AlN, SiC)]
- TiN/MgO界面系、超格子系の計算も行なっている(上記、参考文献[4][5][6][7][8][9]参照)。
- 最近は、ScN/MgO界面系、超格子系の計算も行なっている(上記、参考文献[10]参照)。
- 2018年9月に、SSDM2018で、TiN dot/MgO超格子系のポスター講演を行った。
- 2019年11月27日〜30日、仙台で、ISCSI-8開催。筆者も、TiN(ScN)/MgO関連でポスター講演を行った。
- 2019年12月10日〜14日、横浜で、MRM2019開催。筆者も、TiN/MgO関連でポスター講演を行った。
- 2020年3月1日〜3日、つくばで、MANA INTERNATIONAL SYMPOSIUM 2020 jointly with ICYS(開催中止)。筆者も、TiN(ScN)/MgO関連でポスター講演を行う予定だったが、当該シンポジウムの開催が中止となる。
- 2020年12月10日、11日、ICSPM28がオンラインで開催される。筆者も、TiN(ScN)/MgO関連でポスター講演を行った。
関連有用リンク
(筆者の他の研究)
[10H-BN][6H-AlN][5H-BN][h-BN](JPSJへ)
[Top][Backup]
{kind=link}