[Information on BN (Boron Nitride) compounds][BN][SiC][AlN][Top][English]
(Start, 1/29, 2009)(Update, 6/5, 2026)
- 目次
GaN:窒化物、3C-GaN(ZB-GaN、閃亜鉛鉱構造)や2H-GaN(w-GaN、ウルツ鉱
構造)
祝ノーベル物理学賞(2014年)赤崎勇先生、
天野浩先生、中村修二先生(米国籍)。
2H-GaN(ウルツ鉱構造)の[バンド構造1(png画像、26.6 kb、3d非考慮)][バンド構造2(png画像、28.2 kb、3d考慮)]
筆者の計算では、3d非考慮の2H-GaNでは、平衡格子定数が約2%短めとなる。一方、3d考慮の2H-GaNでは、2%ほど伸びめとなる。
Ga(ガリウム)は浅い3d軌道を持つ。Gaの[バンド構造(png画像、8 kb、3d非考慮、fcc構造)][バンド構造(png画像、7 kb、3d考慮、fcc構造)][関連記事]。
関連文献[Ga]:K. Takemura, K. Kobayashi and M. Arai, Phys. Rev. B58, 2482-2486(1999)
N(窒素)は、大気の約80%を占める気体。いろいろな元素と化合して窒化物を作る。参考:[遷移金属炭化物、窒化物表面関連情報]
窒化物に関しては、本ページでもBNやAlNを扱っている。
GaとNで二元の化合物を形成するとした場合、ウルツ鉱構造(2H-GaN)、閃亜鉛鉱構造(3C-GaN)、岩塩構造などを考えることが出来る。
ウルツ鉱構造と閃亜鉛鉱構造を比べると、第一原理計算ではウルツ鉱構造の方が安定になる。但しエネルギー差は、10 meV/1H-GaN程度と小さい。1H-GaN:Ga1個、N1個の対当たり。
GaNの計算例(文献)。
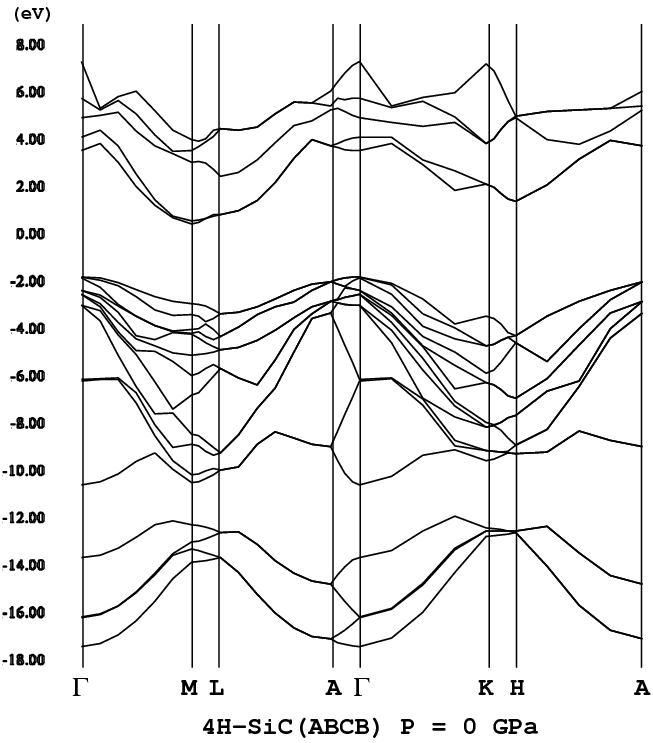
過去、青色の候補として、ZnSeやSiCが挙がっていた。4H-SiCの[バンド構造](png画像、36 kb)、ZnSe(閃亜鉛鉱構造)の[バンド構造](png画像、10 kb)
JSTの2014年ノーベル物理学賞:青色発光LED研究特集ページ(*)
日本物理学会のリリース記事ページ
応用物理学会の特集記事ページ
機構(NIMS)のプレスリリース記事(”「天野 - 小出共同研究ラボ」開所式典及び天野浩教授 (NIMS特別フェロー)記念講演会を開催”)
以上、多少”我田引水”的であることをお断りしておく。
(所感)
筆者自身は、GaNそのものの計算による論文等の報告は行なっていない。関連するIII-V化合物(主にAlNなど)に関する研究例はいくつかある([参考文献]参照)。GaNは、AlNなどの類似系として計算した経験があった。これは、バンド構造図集に載せるために、いくつかのIII-V化合物(BN、AlN、AlSb、GaAs、GaSbなど)のバンド計算を行なうついで程度の感じで計算しただけだった。
GaNにおいて、Gaは上記で示したように比較的浅い3d軌道を持つので、窒素との化合物を形成した場合、この浅い3d軌道の影響が無視できない可能性が高い(と思われている)。実際の2H-GaNのバンド構造を見ると、Gaの3d軌道は、フェルミレベルから14 eVほど下(とそれ以下)に存在する。フェルミレベルから1 Ry程度以上下なので、実際の物性に大きな影響を与えるかは微妙なところである(ただ筆者の計算では平衡格子定数が2%ほど大きいことを考慮する必要があるし、筆者自身もここら辺はよく分からない)。3d考慮と非考慮で、フェルミレベル近傍のバンド構造は、互いに良く似ている(ここでも格子定数の違いによる差〔バンドギャップの大きさなど〕が存在する)。
3d考慮の2H-GaNのバンド構造では、Nの2sバンド(と思われる。←フェルミレベル下約12 〜 14 eV辺り)と3dバンド(と思われる)が重なって(混じり合って)いる。そもそも、これらのバンドが、本当にNの2sか、Gaの3dかは、もっとちゃんと検証して確かめる必要がある。検証の仕方は、いくつか考えられるが、(1)当該バンドの(部分)電荷密度の分布と原子位置の比較、(2)部分状態密度(個々の原子毎のs, p ,d別)の解析などから判定可能である。勿論、s, p, d各バンドは、お互い混じり合っているので、純粋な意味でs, p, dに分けて考えることは出来ない。s的なもの、p的なもの、d的なものくらいに考えて欲しい。
当初、青色発光ダイオードの候補としては、ZnSeやSiCが有力なものとして挙がっていた。既にそれらのバンド構造も上で示している(構造は、ZB-ZnSe, 4H-SiC、ZB:閃亜鉛鉱構造)。ただ、バンドギャップの大きさは、GaNの場合も含めて実験値より過小な値となっている(参考:バンドギャップの過小評価)。
実際に発光ダイオードとして発光させるためには、不純物などをドープして、n型、p型の半導体としなければならない。そこら辺の動作原理に関しては、適宜”固体物理”、”半導体工学”系の書籍(テキスト)を参照されることを薦める。因みに、発光ダイオードの逆の現象が、太陽電池である。バンド間の電子の遷移の仕方で、発光(光の放出)になったり、逆に光を吸収して電気になったりする。
更に、我田引水的だが、今回の物理学賞の話(青色発光ダイオード)は、材料・物性・物質ど真中の内容であり、我々物質・材料(材料・物性)分野の者(本当は物理屋なのだが)にとって、”うってつけ”の話だったりする。このような便乗的な姿勢は、ちょっと情けない感があるが、とにかく乗っておいて損はないだろう。正直、これが材料・物性分野への追い風になってくれると大変有難い。その割には、筆者の周りも、材料・物性分野を見ても、あまり目立った動きが見られないような気がするのは、筆者の気のせいだろうか?。
一日一日こつこつと書き足している。積木のような文章の付加の仕方。GaNは、AlNと比べバンドギャップが小さい(実験値:3.503 eV〔参考文献[N1]:Springer Handbook of Condensed Matter and Materials Data, ed. W. Martienssen and H. Warlimont (Springer, 2005)〕)。AlNのバンドギャップは、6.19 eV(参考文献[N1]に同じ)である。GaNやZnSeのバンドギャップは直接遷移型である。一方、4H-SiCは間接遷移型である。直接遷移(価電子帯の頂上と伝導帯の底が同じk点上に存在)、間接遷移(価電子帯の頂上と伝導帯の底が異なるk点上に存在)の詳細に関しては”固体物理”、”半導体工学”系の教科書を参照。SiCは多数のポリタイプ構造(本ページ上〔ポリタイプとは何か?以下〕参照)が存在するが、少なくとも筆者の知る限り全ての構造でそのバンドギャップは間接遷移型である。発光を考える場合、間接より直接の方が(余計なフォノンの関与が必要ない分)有利である。参考:[(直接ギャップ、間接ギャップ)]
GaNは、III-V化合物の一つで、それらを合成する方法としては、エピタキシャル成長法や、気相成長法を使う。但し、筆者は実験の話には全く明るくないので、これ以上の詳細を語るための材料を持ち合わせていない。ただ、何らかの基盤(物質)の上に、今述べた方法等により、GaNを成長させる。既に多くのところで語られているように、良い結晶を成長させることは、そう簡単な話ではなかったことが窺われる。III-V化合物は、既に述べたAlN(窒化アルミニウム)や、本ページで扱っているBN(窒化ほう素)など、B, Al, Ga, In(III族元素)とN, P, As, Sb(V族元素)との化合物(通常組成比1:1)のことである。因みに、SiC、SiGeなどは、IV-IV化合物、ZnO、CdSなどは、II-VI化合物である。
重ねて述べるが、筆者自身は、GaNのみを対象にした研究で論文は出していない(AlNならある[3][6][7][11][12]←文献[12]は、AlBN関連)。Ga単独の論文は共著で出している(関連文献[Ga])。窒素は、(常温常圧下で)ごく普通の気体なので、これ単独を対象とした研究は行なったことはない(筆者とは無関係な高圧下での固体状態に関する研究例などはある)。
応用物理学会誌「応用物理」、第83巻、第11号(2014年11月号、873頁〜879頁)に、2014年ノーベル物理学賞関連の記事あり。
一方、日本物理学会誌、2014年11月号(第69巻、第11号)には、ノーベル賞関連の記事はない。
関連記事[ページ](←既にアクセス不能。”大学がノーベル賞機に科学政策見直し要望”、サイエンスポータル[JST]の記事)を発見。まあいろいろ”物質・材料関連・基礎科学関連”で追い風になっている(?)。
InGaN, InNに関しての参考論文: Y. Kangawa, T. Ito, A. Koukitsu and K. Kakimoto, Japanese Journal of Applied Physics 53, 100202(2014)[Theoretical approach][InGaN][InN][Epitaxy][Structural stability]
日本物理学会誌、2014年12月号(第69巻、第12号)に、”学会ニュース”記事「2014年ノーベル物理学賞に赤崎勇、天野浩、中村修二の3氏」(佐藤勝昭、884〜885頁)が掲載される。
応用物理学会誌「応用物理」、第84巻、第1号(2015年1月号、巻頭特別企画)、”2014年ノーベル物理学賞受賞者手記”、”ノーベルウィーク報告”、基礎講座:”今さら聞けない?若手会員のためのLED基礎”(菊池昭彦、岸野克巳、66頁)が掲載される。
(講演会情報1)2015年応用物理学会春季学術講演会(東海大学、神奈川)にて、ノーベル賞受賞記念講演会が開催(2015年3月13日、東海大学湘南キャンパス2号館大ホール(2S-101教室))。詳細は案内ページ参照[終了]。
(講演会情報2)The 6th International Symposium on Growth of III-Nitrides (ISGN-6)、11月8日〜13日(2015)、開催地:浜松(静岡)、Nobel Prize Session and Reception(11月9日)あり[終了]。
日本物理学会(第70回年次大会、早稲田大学)でもご講演(天野先生)の予定あり。ssdm 2015(2015年9月27日〜30日、札幌コンベンションセンター)でも天野先生によるPlenary sessionあり[終了]。
日本表面科学会平成27年度特別講演会「世界が照らすLED」、5月23日(2015)午後3時半より、講演者:天野先生、開催地:学習院大学(東京)、詳細は案内ページ参照[終了]。
(講演会情報3)2015年応用物理学会秋季学術講演会(名古屋国際会議場、愛知)にて、特別シンポジウム(天野先生、益川先生)開催(2015年9月13日、名古屋国際会議場センチュリーホール)。詳細は案内ページ参照[終了]。
GaN研究コンソーシアムの設立について:プレスリリースページ(物質・材料研究機構)
(講演会情報4)公開シンポジウム「省エネルギー社会の実現に資する次世代半導体研究開発」、2016年5月18日、開催地:学術総合センター一つ橋講堂(千代田区、東京)。詳細は案内ページ参照[終了]。
(講演会情報5)特別講演会:「10年後の未来から、今できることを考える」、10月20日(2018)午後1時半より、開催地:ES総合館1階ESホール(名古屋、愛知)、詳細は案内ページ参照[終了]。(続く)
第13回NIMSフォーラム:10月24日(2013)、開催地:東京国際フォーラム
(有楽町、東京)[終了][開催報告]にて、”BN, SiC, AlN層状化合物の物性を電子状態計算で探る”という題目でポスター発表した。【参考:公開版】イブニングセミナー発表用原稿:PDF版(PDF形式ページ、約3 MB)
上記関連:バンド計算関連の単位表(png形式、220 kb、NIMSフォーラム版[試作版])(10/23、2013、修正←電流:6.6233 → 6.6236)
NIMSフォーラム当日、上記表の紙版配布(深く感謝)。
↑表内数値等に誤字、間違い等があれば、ご指摘いただけると大変ありがたいです。
筆者は、2010年12月24日に、「理論・シミュレーションによるより
深い材料特性の理解: 第一原理計算によるBNポリタイプと関連物質の研究」
という題目で講演を行なう予定である。ちょうど、BNポリタイプの話が中心と
なるので、本ページでその準備過程を披露する形で、ページ自身の充実を図っ
ていこうと思う。
(12/27、2010)当該セミナーは無事終了。聴講された方々及び準
備等を含めた関係職員の方々に深く感謝。
(【公開版】セミナー発表用原稿:PDF版、約3 MB)
PDF(PDF形式、約3MB):これは公開版です。このためセミナー以外で未発表の部分などを削除して公開しています。内容に関して誤字、脱字、間違い等があるかもしれません(無保証)。ご注意下さい。
(注意)イブニングセミナーに関しては、上記リンク先の案内を参
照して欲しい。残念ながら受講者は既に決定していて、新規の参加が可能かど
うかは、当該ページにある事務局へ問い合わせて欲しい。
「ポリタイプは、1次元に関しての多形構造(ポリモルフ)のことである。」
と言っても、さっぱり分からないだろうと思う。そもそも多形構造と
は何だろうか?。まずは、これについて説明してみる。多形構造は、同じ化学
組成を持つ化合物が、条件によって様々な結晶構造を持ち得ることである。単
体元素でも同様なことが起こり得る。例えば、化合物ならSiO2
(シリカ化合物)は、多くの多形構造を持つ。単体の元素では、炭素がダイヤ
モンド、グラファイトという2つの結晶構造が存在する。広く考えれば、フラー
レンやカーボンナノチューブ、グラフェンなども多形の一種と考えることが出
来る(ダイヤモンドやグラファイト、フラーレンなどは”同素体”でもある)。
SiO2や炭素の同素体の場合は、それぞれ互いの結晶構造が3次
元的に異なっている。一方、ポリタイプは結晶構造の差が1次元方向に限定さ
れたものである。特に、ここで扱うポリタイプは、六方晶層状構造のもののみ
とする。また物質としては、BN、SiC、AlNの3つの2元化合物を計算対象とす
る。特に、SiCは多数のポリタイプ構造を持つことで有名である(A. R. Verma and P. Krishna〔敬称略〕の教
科書参照)。六方晶でないポリタイプは存在する。また、BN、SiC、AlN以外の
物質でもポリタイプ構造を形成するものは多数存在する(が、ここでは扱わな
い)。
詳細に関しては、A. R. Verma and
P. Krishna(敬称略)による教科書、International Table for
Crystallographyや、有用なウェブページな
どを参照して欲しい。
以下、簡単に説明を行なう。説明では誤記や間違った記述、曲解してい
る部分があるかもしれないので注意して欲しい(無保証)。ここ
では、六方晶層状のポリタイプのみを取扱う。このポリタイプは、
sp3結合による四面体構造を成している。BNポリタイプでは、四面
体構造の中心にB原子があるとすると、その周りの最近接原子(四面体なので
4つ存在←理想構造を想定)がNとなる。B層とN層で一つの対と考え、これを
一つの単位と考えて層状構造を表現する。例えば、筆者が最初に扱った5H-BN
構造[1][2]は、B層、N層の対(B-N層)5つからなる六方晶(Hexagonal)なポリ
タイプ構造であることを意味する。で、これらのB-N層の積層の仕方を、表現
する方法がいくつか存在する。
(ABC記法)
六方晶の構造の層で原子の取り得る位置は、A、B、Cの3つの位置しかな
い。A、B、Cは、それぞれポリタイプ構造の単位胞における菱形の平面上の
(0,0),(1/3,1/3),(2/3,2/3)の位置に対応する。(注)座標の取り方によって、
(1/3,1/3),(2/3,2/3)は、(1/3,2/3),(2/3,1/3)と表現される場合がある。加え
て、ここではz軸(c軸)の座標は省略した。ポリタイプの持つ対称性からこ
れ以外の位置に原子は存在することが出来ない。先に示した5H-BN構造の場合、
ABC記法では、"ABCBC"と表現される。"ABCAB"、"ABCAC"という表記も可能
であるが、これらはどれも同じ構造である。実は、5Hポリタイプ構造の取り得
る構造は、この一つしかない。つまり5Hと言った段階で、ユニーク(唯一)に
決まってしまう。同様に、2Hポリタイプは、"AB"、3Hポリタイプは、"ABC"
(3Hポリタイプはちょっと特殊な構造→後述)、4Hは、"ABCB"と全てそれぞれ
一つの構造しかない。ABCは、BCAとしても、ACBとしても、CABとしても、CBA
としても、、、、、、と、どのように並べ替えても同じ構造である。但し、こ
の時、"AA"、"BB"、"CC"のような連続した並びは起こらないものとする。
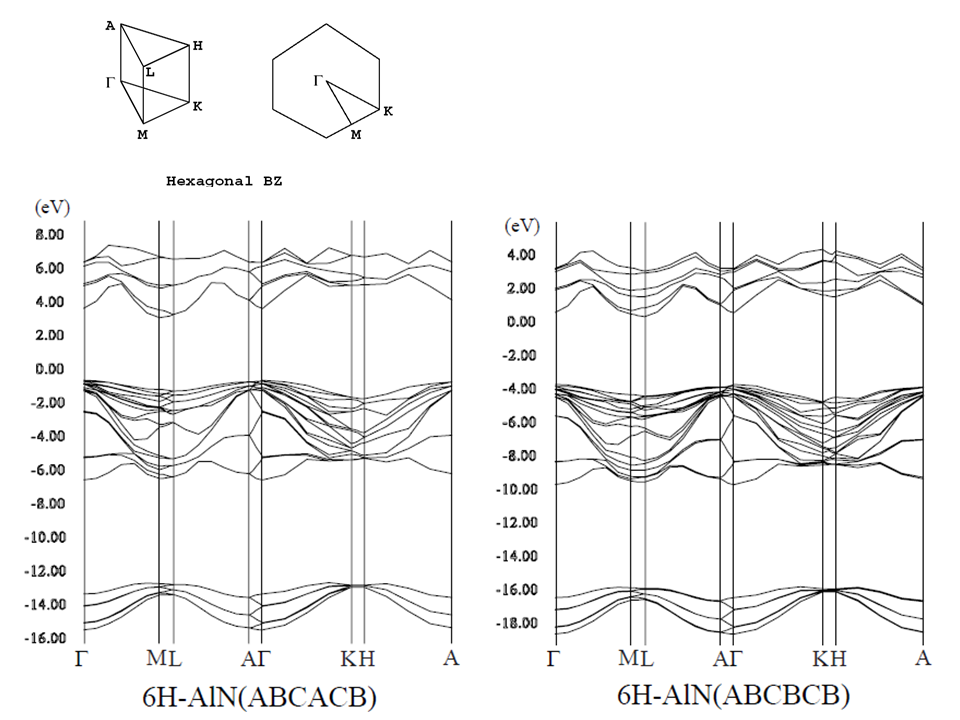
6Hポリタイプは、"ABCACB"、"ABCBCB"の2つの異なる積層構造が存在する。
6H以降のポリタイプが、どのくらいの数の構造を持つかは文献[Ig]を参照して
欲しい。7Hポリタイプで3つ、8Hポリタイプで6つ、9Hポリタイプで9、10H
ポリタイプで18、12Hポリタイプで58、30Hポリタイプで600万の構造が
存在する。nHでのnの値が大きくなるほど、急激に可能な構造の数が増えてい
く。この数は、既に述べたように、BN、SiC、AlNのような2元化合物によるポ
リタイプの場合である。nが大きな値ほど沢山の構造が存在し得るが、それら
の構造のとり得る対称性は、P63mcか
P3m1の2つしかない。
(Hägg記法)
ポリタイプの積層の仕方を記述する方法は、ABC記法だけではない。他に
もいくつか記述の仕方が存在する。重要なのは、それぞれの記法から他の記法
への書き替えが可能であることである。ここでは、Hägg記法について説
明する。ABC記法において、A→B→C→A(の繰り返し)とA→C→B→
A(の繰り返し)の2つの繰り返しパターンに分けることが出来る。前者の場
合を”+”、後者の場合を”ー”と表現するのが、Hägg記法である。例
えば、|ABC|ABC|ABC|・・・は("|ABC|"は一つの周期的単位とする)、
|+++|+++|+++|・・・となり、|ACB|ACB|ACB|・・・は、|---|---|---|・・・
となる。(注)但し、ABCとACBは、構造としては同じものである。
(h-c記法)
h-k記法とも書く(実はこちらの”言い方”の方が多い)。また、
"Jagodzinski記法"とも言う。先の、Hägg記法において、"++"または"--"
が、"c"(cubic)、"+-"または"-+"が、"h"(hexagonal)に対応するとして、ポリ
タイプの積層を表現した記法である。例えば、4Hポリタイプは、Hägg表
示で、"++--"なので、そのh-c表示は、"hchc"(別に、"chch"としても問題は
ない)となる。6Hポリタイプでは、"+++---"と"++-+--"の2つの異なる構造が
あり、それぞれ、"hcchcc"、"hchhhc"となる。"hchhhc"は、"hhhchc"でも
"hchchh"としても一向にかまわない(周期性を壊さない限り区切りはどこに設
定しても問題ない)。ここでのh-c記法の書き方は標準的でないかもしれな
い(調査中)。ここで、一周期での"h"と"c"の総数pと、一周期での"h"の
数n_hの比、n_h/pが、"hexagonality"(H)である。
Hexagonalityは、ポリタイプを考える上で、大変重要な量(パラメー
タ)である。
(Zhdanov記法)
ポリタイプにおいて、Hägg記法によって表記された"+"、"-"の数(同
一の記号が連続している場合は、その連続した記号の数)の並びによる表記の
方法が、Zhdanov記法である。例えば、4Hポリタイプは、"++--"なので、"22"
となる。6Hポリタイプでは、"+++---"の場合は、"33"、"++-+--"の場合は、
"2112"となる。
(各記法への変換)
あるポリタイプ構造に関して、ABC記法、Hägg記法、h-c記法、
Zhdanov記法のどれか一つの表記が分かっていれば、残った他の全ての記法へ
の変換が可能である。例えば、ABC記法で、ABACABCBCACBと記述されるポリ
タイプ構造(12Hポリタイプ)は、
|ABACABCBCACB|ABACABCBCACB|ABACABCBCACB|...と続いていく。これの他の記
法への変換を以下に示す。
まず、
|ABACABCBCACB|A B A C A B C B C A C B|ABACABCBCACB|
- + - - + + + - + + - - -
から、"+ - - + + + - + + - - -"(Hägg記法、一周期分)となる。こ
れから、
- -|+ - - + + + - + + - - -|+ - - + + + - + + - - -|+...
c h h c h c c h h c h c c h h c h c c h h c h c c
となり、"hhchcchhchcc"(h-c記法)を得る(一周期分)。これは、
|h h c h c c h h c h c c|h h c h c c h h c h c c|...
と周期的に並んでいることを意味する(Hexagonality = 50%もこれで分
かる)。また、
|+ - - + + + - + + - - -|
1 2 3 1 2 3
から、"123123"(Zhdanov記法)が得られる。"|"は、周期毎の区切り記号。
(表1)2H,3H,4H,5H,6Hポリタイプに対する各記法での
表記の仕方
| - |
ABC記法 |
Hägg記法 |
h-c記法 |
Zhdanov記法 |
Hexagonality(%) |
| 2H |
AB |
+- |
hh |
11 |
100 |
| 3H |
ABC |
+++ |
ccc |
3 |
0 |
| 4H |
ABCB |
++-- |
hchc |
22 |
50 |
| 5H |
ABCBC |
++++- |
hhccc |
41 |
40 |
| 6H |
ABCACB |
+++--- |
hcchcc |
33 |
33.3 |
| 6H |
ABCBCB |
++-+-- |
hchhhc |
2112 |
66.7 |
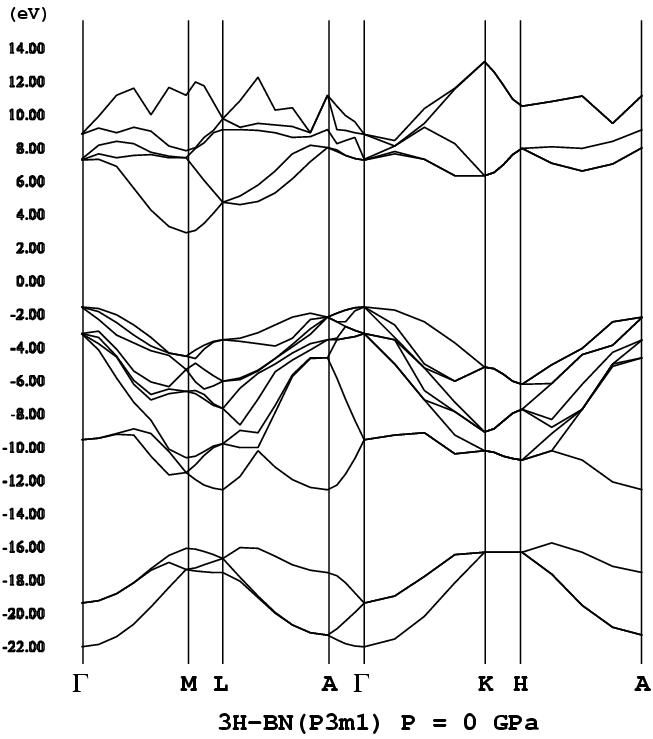
(注)3Hは、3C(立方晶構造〔閃亜鉛鉱構造〕)をポリタイプとして考えた
もの。3C(3H)の、Zhdanov表記は(ここでは"3"としたが)、"∞"とされること
が多い。
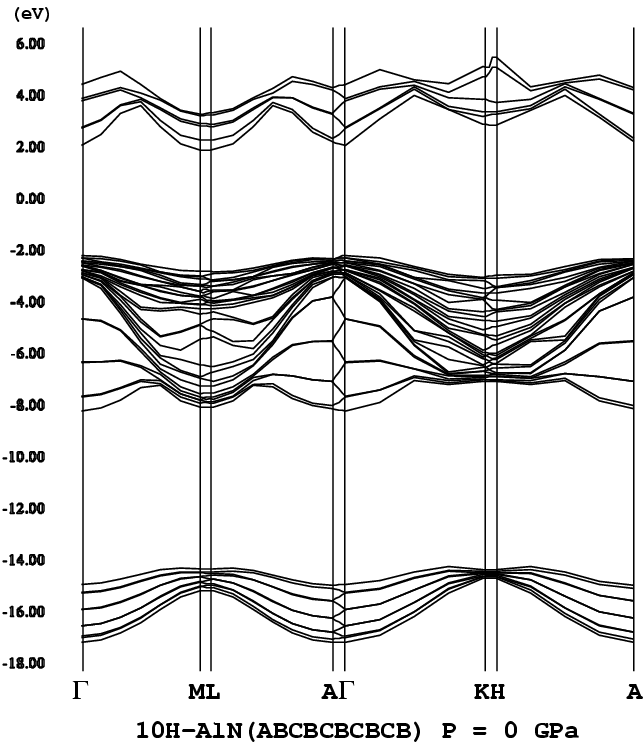
10Hポリタイプに関しては、文献[6]の表1(Table I)を参照して欲しい。
Hägg記法以外の各記法に対応した、10Hポリタイプでの可能な全構造(1
8個ある)の一覧となっている。
Zhdanov表記(2H 〜 9H)
2H : 11
3H(=3C) : ∞
4H : 22
5H : 41
6H : 33
6H : 2211
7H : 52
7H : 4111
7H : 3121
8H : 71
8H : 44
8H : 3212
8H : 3311
8H : 221111
8H : 211211
9H : 63
9H : 5211
9H : 5112
9H : 4221
9H : 4122
9H : 3231
9H : 312111
9H : 311121
9H : 411111
(以下、変換例)
"311121" <-> "+++-+-++-" <-> "hcchhhhch" <-> "ABCACACAB"
Hexagonality("311121", 9H) = 6/9 = 2/3 = 66.7 %
1999年、小松(無機材質研究所〔当時〕、〔現〕物質・材料研究機構、
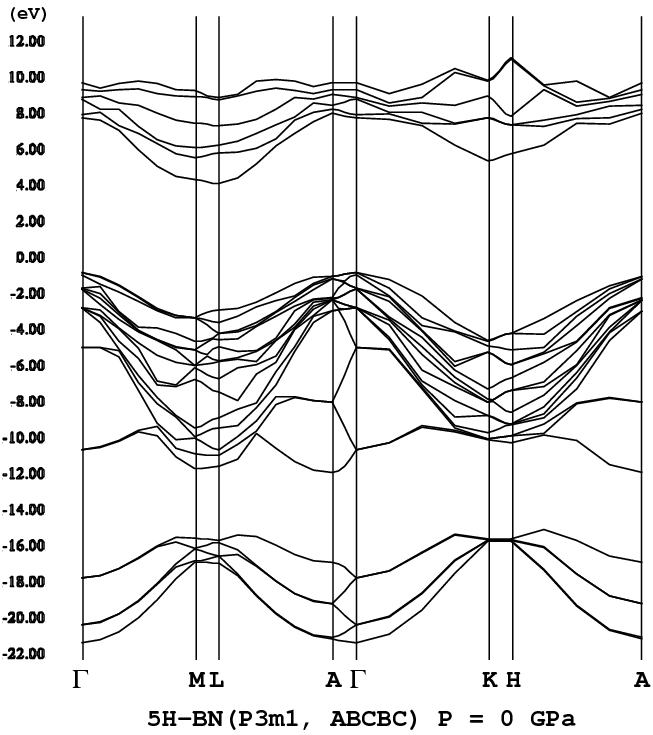
半導体材料センター)等によって5H-BNが世界で初めて合成された[1]。5H-BN
は、5Hポリタイプ構造を持ったBN(窒化ホウ素)のことである。合成手法は、
プラズマCVD法である(CVD:化学気相成長法)。この5H-BNの電子状態(電子
構造)を第一原理計算手法を用いて筆者等は求めた[2]。5H-BNは、既に表1で
示した5Hポリタイプ構造であり、sp3結合による四面体構造を基本
構成要素としている。既に述べたように、5Hポリタイプで可能な構造は一つし
かない。その構造の持つ対称性は、P3m1であり、Hexagonality
は、40%である(B-N層5層の内、2層がhexagonalな層となっている。従っ
て、2/5=40%)。5H-BNの構造に関しては、文献[2]の図1を参照して欲
しい。
5H-BNの計算の後、文献[3][6][7][9]とBNと関連する化合物(SiC, AlN)のポ
リタイプ構造の計算を続けた。扱ったポリタイプは、2H、3H(3C)、4H、6H、
10H、12H、30Hである。5Hまではそれぞれ一つの構造がユニーク(唯一)に決
まるが、6H以降は複数の可能な構造が存在する。6Hポリタイプは、2つしか可
能な構造が存在しないので全ての構造で計算可能だったが、10Hでは可能な構
造は18にもなる(12Hでは58存在)。pHのp(整数)の数が増えることは、
計算として扱うべき原子数が増えることを意味する。通常の第一原理計算では、
原子数の3乗で計算量が増えるので10H、12Hポリタイプ構造の計算は、2H、4H、
6Hポリタイプの計算と比べて、10倍以上の規模になる(6H→12Hで8倍)。
従って、10Hポリタイプでの可能な18個構造全てを計算することは、大変困
難である。12Hなら尚更である。実際には、可能な構造の中からいくつかを”
選択”して計算を行なった。BNポリタイプに関しては、30Hまで計算を行なっ
た[9]。30Hポリタイプには、可能な構造が600万以上存在する[Ig]。当然、
全ての構造を尽くして計算することは不可能であり、30Hポリタイプの中で最
もHexagonalityが小さい場合(H = 6.7 %)と、最も大きい場合(H = 93.3 %)
から各一つずつ構造を選んで計算した。
ここで筆者が使用している計算手法について述べてみる。
筆者が使用している手法は、平面波基底+ノルム保存型擬ポテンシャルを使
用した、第一原理分子動力学手法(広い意味でのカー・パリネロ法
[CP]、Car-Parrinello法)である。平面波基底は、電子の波動関数を記述する
基底関数である。擬ポテンシャルは、物性に直接関わる価電子部分のみからな
るポテンシャルである。従って、内殻電子からの寄与は通常無視する。この擬
ポテンシャルの基で、固体内の電子状態を数値的に解く方法の一つが、バンド
計算である。特に実験結果に何ら依らないバンド計算のことを第一原理バンド
計算と言ったりする。”第一原理”とは、既に述べた、「何ら実験結
果に依らない」という意味である。類似な言い方として、非経験的、
ab-initioなどがある。
第一原理分子動力学計算は、電子が計算対象である第一原理バンド計算にお
いて、格子構造の最適化、つまり構造の安定化(或いは分子動力学計算)まで
考慮した手法である。これは、1985年に出現したカー・パリネロ法[CP]が
(第一原理分子動力学手法の)始まりであると言っていいだろう。
第一原理分子動力学手法、擬ポテンシャル、平面波基底、バンド計算、電子
状態の計算において重要な密度汎関数理論、局所密度近似などのより詳細な解
説として、「徹底
解剖 第一原理計算」の支援ページにある、筆者の連載記事(雑誌「金属」
で連載)を挙げておく。勿論、当該連載で更により詳細或いは有用な文献等の
情報も紹介されている。
尚、ここで扱っている第一原理計算では、温度は絶対零度(T = 0 K)を前提
として行なわれている。局所密度近似の型は、von Barth and Hedinのもの
[BH]を使用している(一部、Wigner[Wigner]のものを使用したものあり)。擬
ポテンシャルは、ノルム保存型のTM型[TM]を使用(参照:NCPS2K)。
Siに関しては、BHS型[BHS]を使用。Alには、部分内殻補正[PCC](原子の内殻
部分の寄与を部分的に考慮するための補正近似)を考慮している。擬ポテンシャ
ルの非局所部分に対して、KB近似[KB](実際の計算量を大幅に軽減させる)を
導入している。
さてここでは、これまでのポリタイプ構造に関しての計算結果について説明
していく。
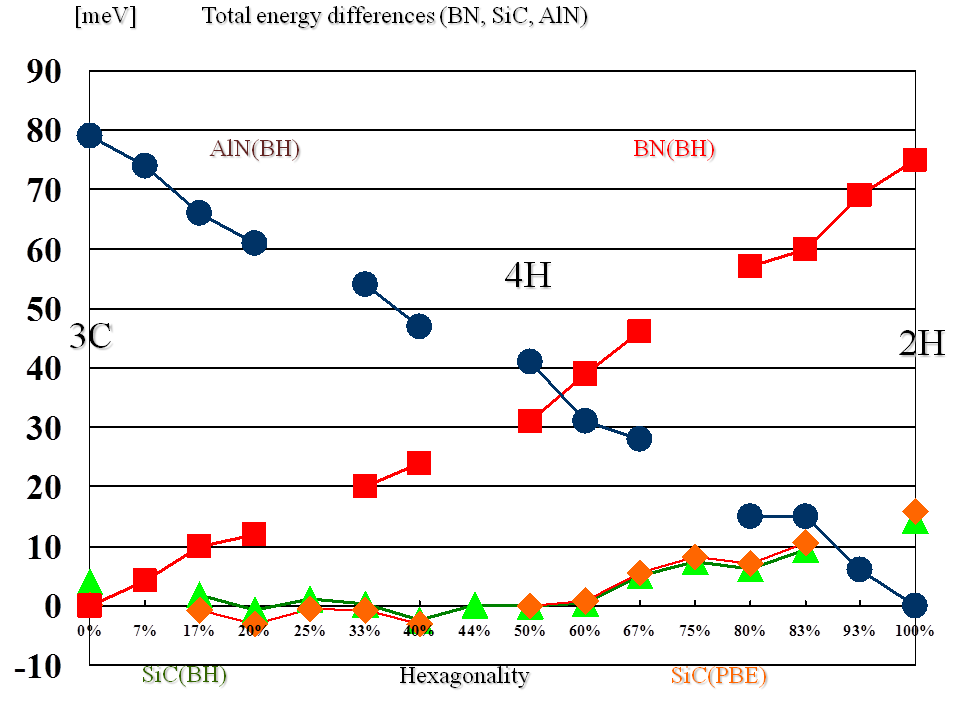
(全エネルギーとHexagonalityとの関係)これまでの計算から、BN
ポリタイプとAlNポリタイプに関して、それらの(計算によって得られた)全
エネルギーとHexagonalityとの間には深い関係があることが分かっている。全
エネルギーは、計算によって得られる扱った系(ポリタイプ構造)の総エネル
ギーであり、絶対零度での値なので、自由エネルギーにおける内部エネルギー
に相当する(圧力もここではゼロと考えている)。この全エネルギーと
Hexagonalityとの関係は次のようになる。
BNポリタイプにおいては、Hexagonalityが小さいほど全エネルギー
が低くなる。
一方、AlNポリタイプでは、Hexagonalityが大きいほど全エネル
ギーが低くなる。
全エネルギーがより低いということは、より構造として安定と考えてもらっ
てかまわない(但し、絶対零度で圧力ゼロを想定)。つまり、BNポリタイプで
は、H = 0 %の3C-BN(3H-BN)が最も安定であり、AlNポリタイプでは、H = 100
%の2H-AlNが最も安定ということになる。筆者のこれまでの計算で、BNでは、
2H、3H(3C)、4H、5H、6H、10H、12H、30Hポリタイプ、AlNでは、2H、3H(3C)、
4H、5H、6H、10Hポリタイプにおいて確認している。尚、3H(3C)ポリタイプ以
外で、H = 0 %となるポリタイプ構造は存在しない。同様に、2Hポリタイプ以
外で、H = 100 %となるポリタイプ構造は存在しない。
全エネルギーとHexagonalityとの関係の具体的な結果については、文献
[2][3][6][7][9]にある表等を参照して欲しい。一方、SiCポリタイプに関して
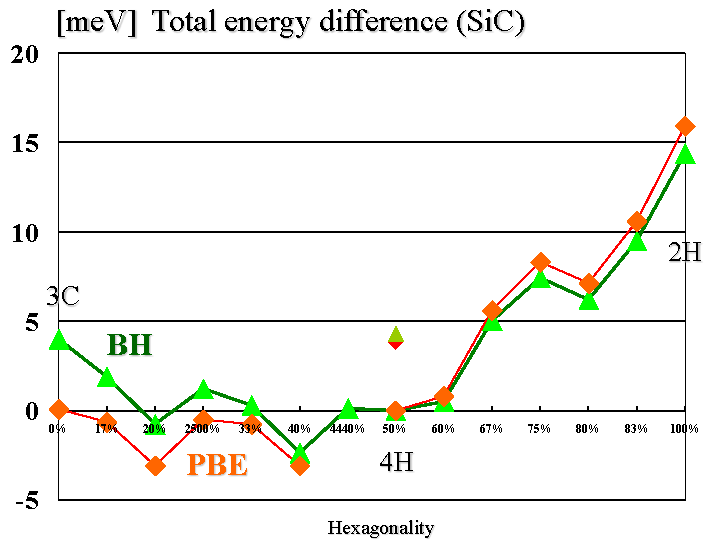
は、全エネルギーとHexagonalityとの間にはっきりとした関係は見られない
[3]。
BNポリタイプ、AlNポリタイプで全エネルギーとHexagonalityに深い関わり
があることは、以下によって説明出来る。それは最短の第三隣接原子の対(こ
の原子対は、必ず陽イオン、陰イオンの対となる)の存在である。ポリタイプ
構造は、四面体構造が基本構造となっていて、理想的な四面体構造では、第一
隣接原子対は4つ、第二隣接原子対は12存在し、それらはそれぞれ原子
対の原子間距離が等しい。等しくなくなるのが第三隣接からで、第三隣接
原子対には、いくつかの種類が存在する。ここで着目するのは、”最短
”の第三隣接原子対である。実際の計算では、構
造最適化(安定構造を求めること)を行なうので、理想的な四面体構造ではな
くなるが、構造の変位(含む単位胞内の原子の変位)は十分に小さいので最短
の第三隣接原子対が、構造最適化によって最短でなくなることはない。
Hexagonalityとこの最短の第三隣接原子対には強い関係がある。
Hexagonalityの定義における、hexagonalな層とcubicな層において、
hexagonalな層(陽イオン、陰イオンの対としての二層)が必ず最短の第三隣
接原子対に対応する。つまりこの最短の第三隣接原子対とHexagonalityは1対
1の対応関係となっており、Hexagonalityが大きいほと、単位胞(周期的境界
条件の基で設定される固体結晶を構成する基本的な単位)内の最短第三隣接原
子対の数は多くなる。
AlNポリタイプにおいて、AlNの結合は共有結合とイオン結合の混ざったもの
となっているが、イオン結合的な度合がより優勢である。従って、AlNポリタ
イプは、最短の第三隣接原子対(陽イオン - 陰イオンの対)のイオン的結合
をより強くして構造の安定を図ろうとする。このため、この第三隣接原子対は
理想位置から、より接近するように変位する。これによってポリタイプ構造全
体としてエネルギー的に得となる。一方、BNポリタイプにおいては、イオン性
と比べて共有結合性が非常に強いので、第三隣接ではなく、第一隣接原子対同
士(この対も陽イオン - 陰イオン対であるが、イオン結合より共有結合がよ
り強く働いている)がより強く結合して安定化を図ろうとする。第一隣接原子
対が近づくと、自動的に最短の第三隣接原子(それが存在すれば。←cubicな
層には存在しない)は離れるように変位する(イオン性が強い場合は、これは
損になる)。共有結合性が強ければ、第三隣接部分で損をしても、第一隣接部
分でエネルギー的により大きな得を得ようとする訳である。この意味で、BNポ
リタイプでは最短の第三隣接原子対がより少ない(=hexagonalな層がより少
ない)方が有利となる。勿論、AlNポリタイプはその逆となる。以上から、
AlNポリタイプでは、Hexagonalityが大きくなる
(最短の第三隣接原子対の数が多い)ほどより安定となり、BNポリタイプでは、
Hexagonalityが小さい(最短の第三隣接原子対の数が少ない)ほどより安定と
なる。
このように、BN、AlN両ポリタイプには、明確な全エネルギーと
Hexagonalityとの関連が存在するが、SiCポリタイプについては、はっきりと
した関係が見えていない。SiCポリタイプは、多くの構造が知られており[VK]、
数多くの実験、理論による研究が行なわれている(文献[3]にある参考文献等
参照)。これまでの理論計算(含む筆者の研究[3])からhexagonalなポリタイ
プでは、4H-SiCが最も安定であった。ただこれは局所密度近似(LDA)によるも
ので、一般化された密度勾配近似(GGA)では、6H-SiC(ABCACB積層の場合)が
最も安定となる[LN]。いずれの場合でも、4Hポリタイプと6Hポリタイプの全エ
ネルギー差は小さい。4H-SiCが最も安定と考えた場合、そのHexagonalityは、
50 %である。過去の研究からそれなりの解釈([PCLC]などいくつかあり)があ
るが、何故4HがSiCポリタイプにおいて一番安定(全エネルギーが低い)かは
明確に説明されていない(と筆者は考える)。これに関して、最近筆
者は新たな結果を得ているが、ここでの言及は控える。
バンド構造に関して
バンド構造に関して、概ねの構造は、BN、SiC、AlN各ポリタイプで類似して
いる。AlNではイオン性も強いが、全体として共有結合によりsp3
結合による、四面体構造を基本とした系なので、お互いのバンド構造が似てい
ることは、そう意外な話ではない。
[具体的な計算結果としてのバンド構造]
上記で示された以外のバンド構造に関しては、文献[2][3][6][7][9]にある
図(バンド構造)を参照。
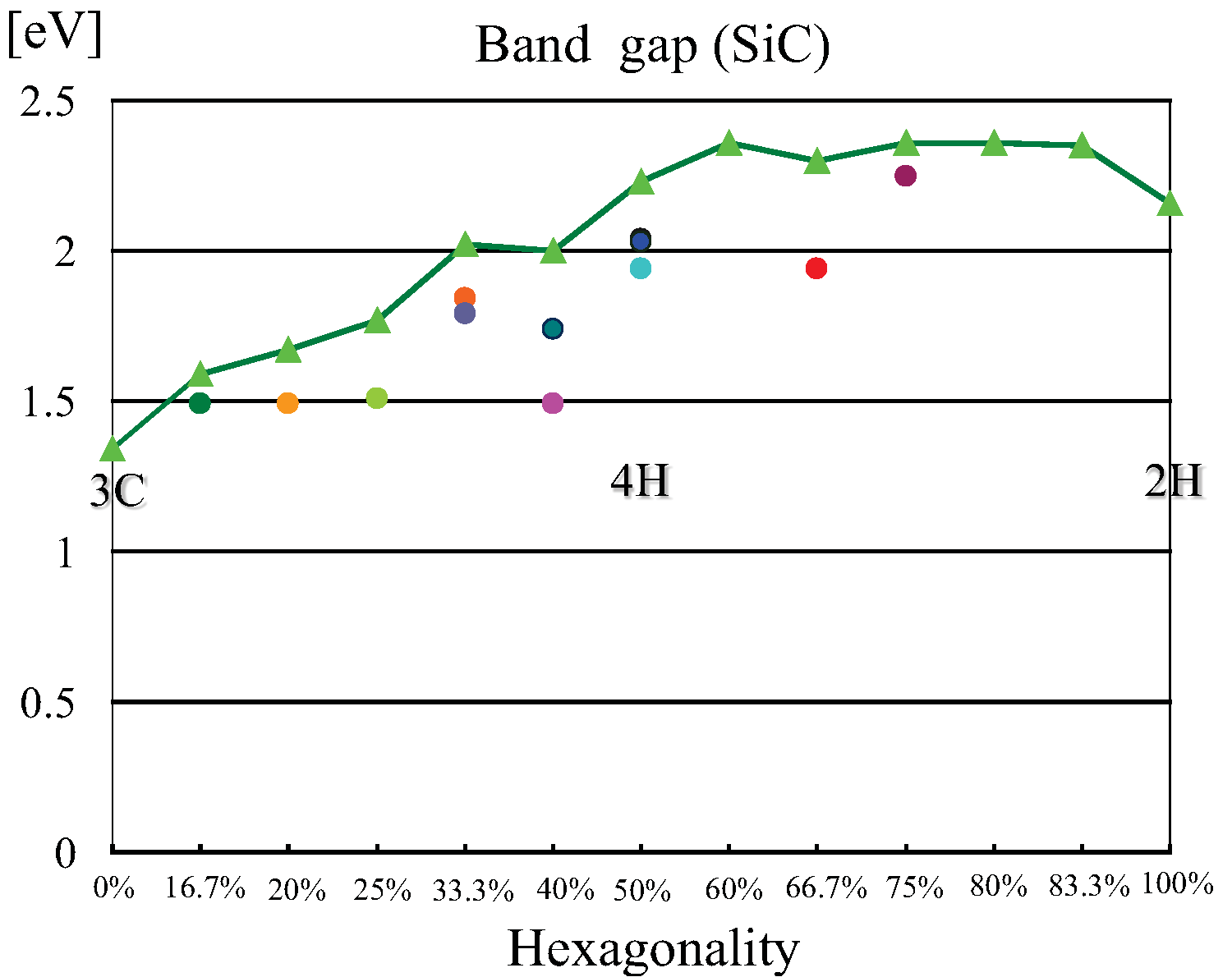
これらのバンド構造から分かることは、全てのバンド構造にはバンドギャッ
プが存在し、これらが半導体(絶縁体)であることを示している。筆者が使用
するバンド計算手法は、局所密度近似を用いているのでバンドギャップの値は、
常に実験値より過小評価される。各ポリタイプのバンドギャップの値は、先に
挙げた文献の表参照。バンドギャップには、間接遷移型と直接遷移
型とがあるが、ほとんどの計算したポリタイプのバンドギャップは、
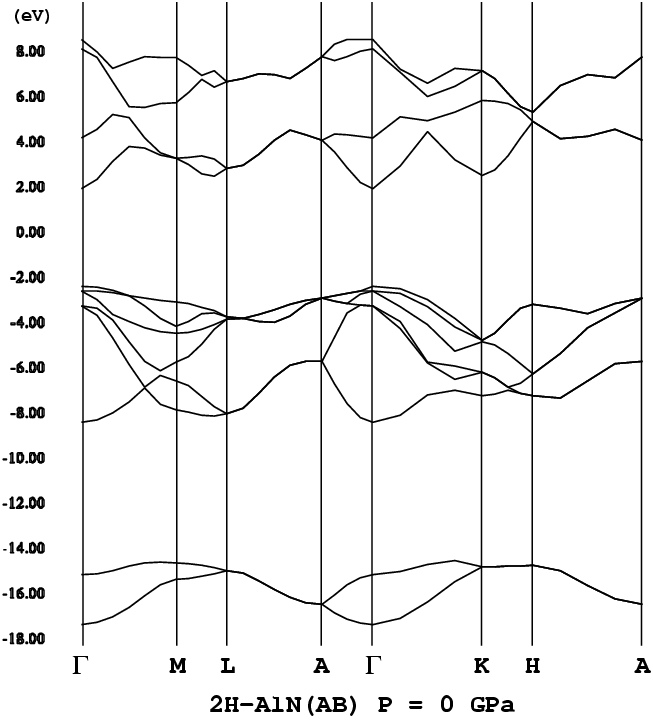
間接遷移型であった。ごく一部(例:2H-AlN)において、バンドギャップ
が直接遷移型であった。AlNポリタイプに関しては、4H-AlN、6H-AlNの第一原
理計算によって得られたバンドギャップが直接遷移型になるという報告
[C1][C2]がある。
筆者等の計算では、4H、6H-AlNポリタイプでは、直接遷移型のバンドギャッ
プにならなかった。ただ、6Hより更に大きな、10Hポリタイプでは直接遷移に
非常に近い間接遷移型のバンドギャップを持つ構造の存在が分かっている[6]。
6H以上のポリタイプでは、複数の構造が存在し、pHにおけるp(整数)が大き
いほど多数の構造が存在する。従って、12H以上(p >= 12)のAlNポリタイプ
において、直接遷移型のバンドギャップとなる構造が存在する可能性はある。
(補足説明)
バンド構造:固体の電子状態(電子構造)を表現する方法の一つ。
逆格子空間における
ブリュアンゾーン上の各k点でのエネルギー固有値(固体内の電子状態を
数値的に解いて得られる電子のエネルギー固有値)を描画したもの。E-k曲線
とか、E-k分散、分散曲線、バンド分散などとも言う。逆格子空間に関しては、
固体物理の教科書等を参照して欲しい。
直接遷移型のバンドギャップ:バンドギャップの空いたバンド構造
において、価電子帯のてっぺん(頂上)と伝導帯の一番底が同じk点であるも
の。k点は既に述べたブリュアンゾーン内の点。関連語:遷移許容、双極子遷
移
間接遷移型のバンドギャップ:直接遷移型でないバンドギャップの
こと。つまり、価電子帯のてっぺん(頂上)と伝導帯の一番底が、それぞれ異
なるk点上に存在する。この場合、電子が遷移するためには、フォノン(格子
の振動を量子化したもの)からの寄与が必要となる。実際のデバイスや発光
(光励起)などへの応用にとって、間接遷移型より、直接遷移型の方がフォノ
ンの介在がない分有利となる場合が多い。
全エネルギー:ここではバンド計算で扱う系の総エネルギーのこと。
詳細は、リンク先の説明参照。絶対零度、圧力ゼロ(T = 0 K, P = 0 GPa)での
全エネルギーは、自由エネルギー(F = E + PV - TS、F:Gibbs自由エネルギー、
E:全エネルギー、PV:圧力×体積、TS:温度×エントロピー、H = E + PV:
エンタルピー)の内部エネルギー(T = P = 0なので自由エネルギー=内部エ
ネルギー。つまり圧力ゼロ、絶対零度での内部エネルギー)に相当する。
他にも筆者は、BN関連の計算を行なっている。文献[4]で筆者等(小林、渡
辺、谷口)は、h-BNと積層をいろいろと変えた類似層状BN化合物の計
算を行なった。h-BNは、上記で扱っているポリタイプと異なり、
sp2結合による層状構造となっており、同様の構造の物質としてグ
ラファイト(石墨、黒鉛)が有名である。
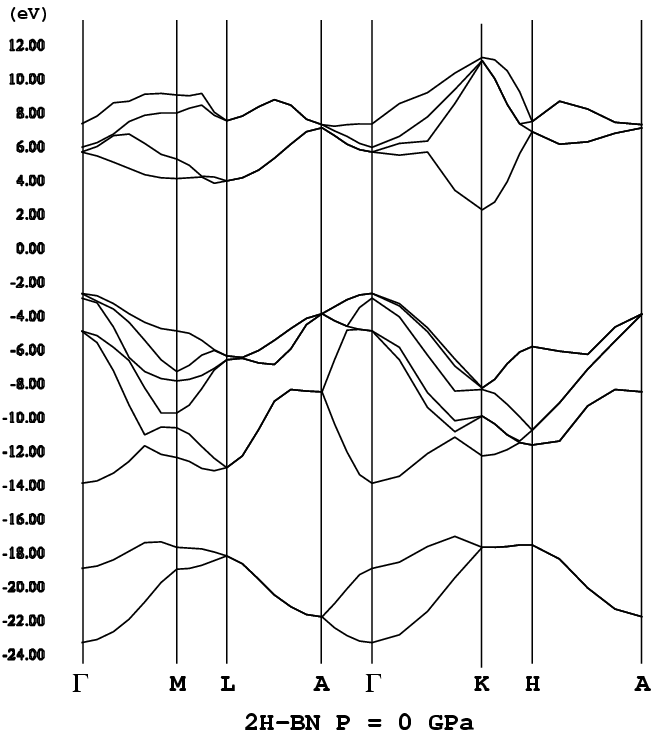
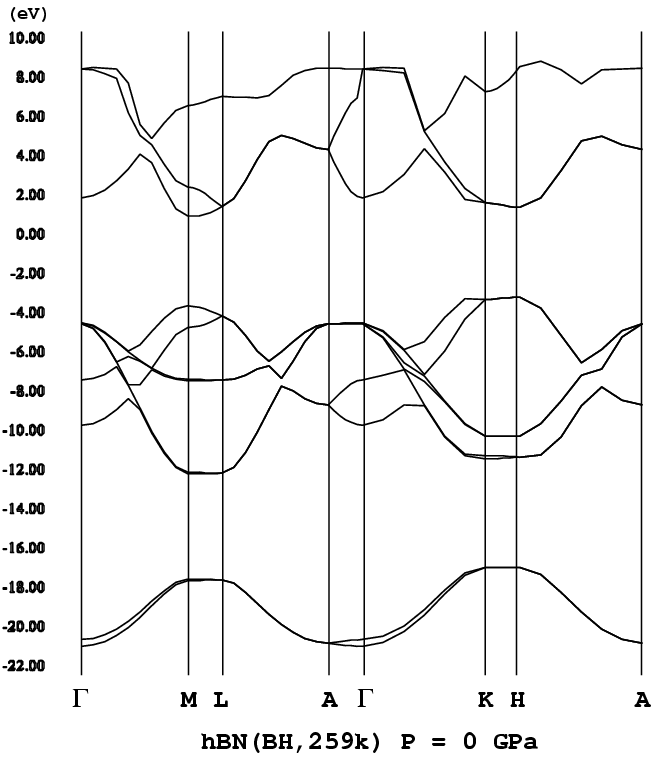
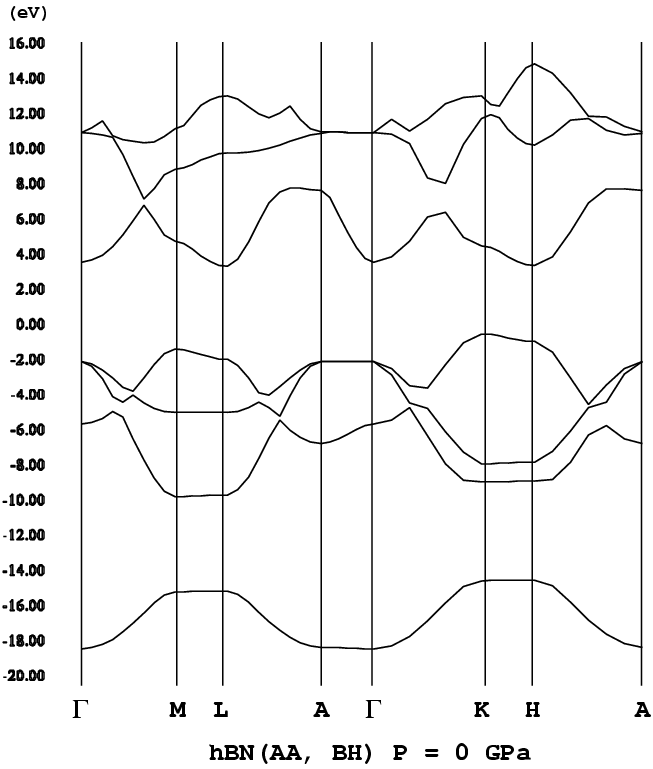
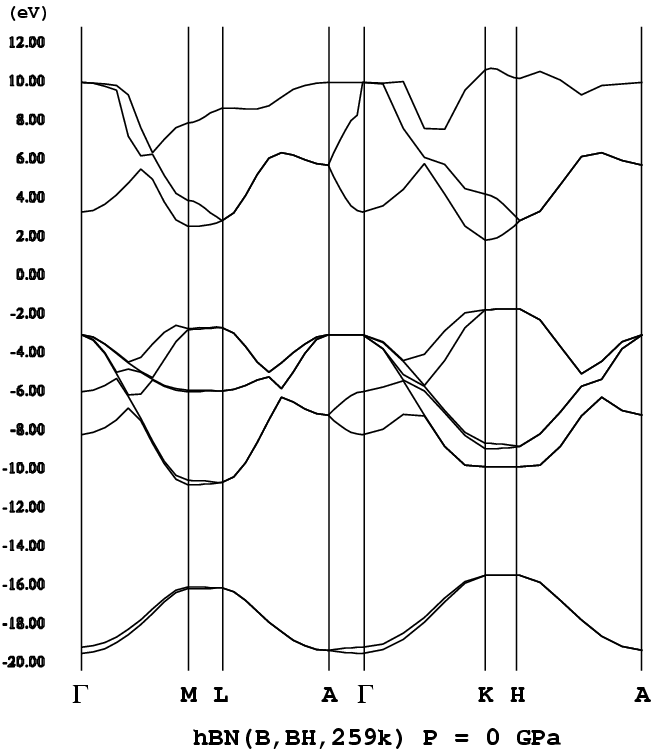
h-BNもsp3結合によるBNポリタイプと同様にワイドギャッ
プ(バンドギャップが大きいこと)な半導体である。ただバンド構造は、
sp3結合によるポリタイプとは大分異なる。特に、h-BNで
はバンドギャップを挟んで価電子帯、伝導帯に平坦なバンドが存在する。ポリ
タイプでもpHのpが大きなものでは、バンドの折り畳みによって平坦なバンド
が生じるが、ここでそれは議論の対象としない。h-BNの
バンド構造:M-L線、K-H線の価電子帯のてっぺん(頂上)部分が平坦になっ
ている。h-BNのバンドギャップは、多くの理論計算(含む筆者による
もの)で間接遷移型を示す。ただ実験[5]によるとh-BNのバンドギャッ
プは直接と観測されている。この不一致についての解釈、検討を行なったのが
文献[4]である(先行研究[LFS]あり)。計算上、h-BNは間接遷移型で
実験との不一致を完全に説明は出来ていないが、一つの解釈としては、AB積層
のh-BN以外にも、エネルギー的に安定ないろいろな積層をした構造を
考えることが出来る。計算してみると、その中には実質上、バンドギャップが
ほぼ直接とみなせるものが存在する。加えて、その直接遷移が遷移許容である
ことも示した(詳細は文献[4]参照)。これには、バンドが平坦なため、各k
点毎のエネルギー固有値の差が非常に小さいことが重要な意味を持つ。
文献[10]で、筆者等は、8H、10H、12H、18H-SiCポリタイプ構造の第一原理
電子状態計算を行なった。SiCポリタイプに関しては、これまで多くの実験、
理論からの研究が行なわれてきた(本ページ上の参考文献、サイトや文献[10]
内の参考文献参照)。これまでの多くの理論計算では、2H、3C、4H、6H-SiCポ
リタイプが計算対象であった(菱面体構造を除く)。8Hより大きなポリタイプ
の計算例、実験例は多くはない。計算上最も安定な構造は、4Hまたは6Hであっ
た(ここでの6Hの積層は、ABCACB)。今回、筆者等の計算で最も安定なポリタ
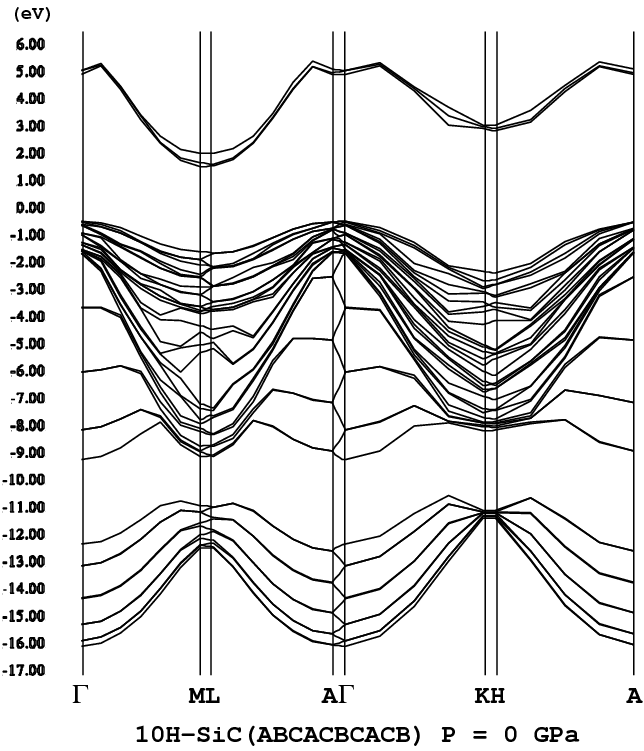
イプ構造は、4H、6Hではなく、10H構造であることが分かった。10Hポリタイプ
構造には、可能な構造が18個存在する(文献[6]参照)。その中の3つの構
造(Zhdanov表示で、3322、55、82)が4H-SiC(筆者等のLDAの計算でも2H、3C、
4H、6Hの中で4Hが最も安定:文献[3])より安定であった。今回の計算でもSiC
ポリタイプでは、全エネルギーとhexagonalityとの間には何の関係も見い出せ
なかった。何故、10H-SiCの中に最も安定なポリタイプ構造が存在するかも解
明されていない。ただ、最も安定な構造は、3322(Zhdanov表示)であり、過
去の研究(文献[10]内の参考文献参照)からZhdanov表示において、"3"、"2"
からなる構造は安定であるということが分かっており、これと整合する(ただ、
次に安定な10H-SiCポリタイプ構造のZhdanov表示は、"55")。同様に、
Zhdanov表示で"1"を含むものは不安定であることが分かっている(これも文献
[10]内の参考文献参照)。今回の計算結果も、一つの例外を除いて概ねこれを
支持するものとなっている。
今回の計算では、LDA(文献[BH])以外にGGA(文献[PBE])による計算も行なっ
た。GGAによる結果は、LDAによる結果と概ね整合しているが、3C-SiCだけが極
端に安定になる(4H-SiCとのエネルギー差は、0.06
meV/Si2C2しかない)という結果が得られた。これが
本当に計算として正しい結果かどうかの結論は出ていない。検証を継続中であ
る。
- [参考文献][文頭]
- [1] S. Komatsu, K. Okada, Y. Shimizu, and Y. Moriyoshi: J. Phys. Chem. B103 (1999) 3289 [5H-BN].
- [2] K. Kobayashi and S. Komatsu: J. Phys. Soc. Jpn. 76 (2007) 113707 [5H-BN].
- [3] K. Kobayashi and S. Komatsu: J. Phys. Soc. Jpn. 77 (2008) 084703 [2H - 6H-BN(AlN, SiC)][6H-AlN][6H-SiC]), and related references therein.
- [4] K. Kobayashi, K. Watanabe, and T. Taniguchi: J. Phys. Soc. Jpn. 76 (2007) 104707 [h-BN].
- [5] K. Watanabe, T. Taniguchi, and H. Kanda: Nat. Mater. 3 (2004) 404 [Experiment for h-BN].
- [6] K. Kobayashi and S. Komatsu: J Phys. Soc. Jpn. 78 (2009) 044706 [10H-BN, 10H-AlN].
- [7] K. Kobayashi and S. Komatsu, "First-Principles Study of 6H-AlN under various pressure conditions", J. Phys.: Conf. Ser. 215, 012111(2010)[AIRAPT22].
- [8] S. Komatsu, K. Kobayashi, Y. Sato, D. Hirano, T. Nakamura, T. Nagata, T. Chikyo, T. Watanabe, T. Takizawa, K. Nakamura, and T. Hashimoto: Journal of Physical Chemistry C 114, 13176 - 13186(2010).
- [9] K. Kobayashi and S. Komatsu: "First-Principles Study of 30H-BN polytypes", Materials Transactions, Vol. 51, No. 9 (2010) 1497[6H-BN, 30H-BN].
- [10] K. Kobayashi and S. Komatsu: "First-Principles Study of 8H-, 10H-, 12H-, and 18H-SiC Polytypes", Journal of the Physical Society of Japan, Vol. 81, No. 2 (2012) 024714[8H-SiC, 10H-SiC, 12H-SiC, 18H-SiC].
- [11] K. Kobayashi and S. Komatsu, "First-Principles Study of Various BN, SiC, and AlN polytypes", Trans. MRS-J, Vol. 37, 583-588 (2012)[go to JST][IUMRS-ICEM2012][48H-BN][20H-SiC][30H-AlN].
- [12] K. Kobayashi and S. Komatsu, "First-Principles Study of AlBN and Related Polytypes", Trans. MRS-J, Vol. 38[3], 485-492 (2013)[4H-AlBN][4H-AlAsN][4H-AlPN][2H-, 3H-, 5H-, 6H-, and 12H-AlBN][3x2H-AlBN].
- [13] No data.
- (他の参考文献)
- [Z] K. Kobayashi: Materials Transactions, Vol. 46, No. 6 (2005) 1094 [Anisotropic compression].
-
- [BNポリタイプなどのバンド構造]
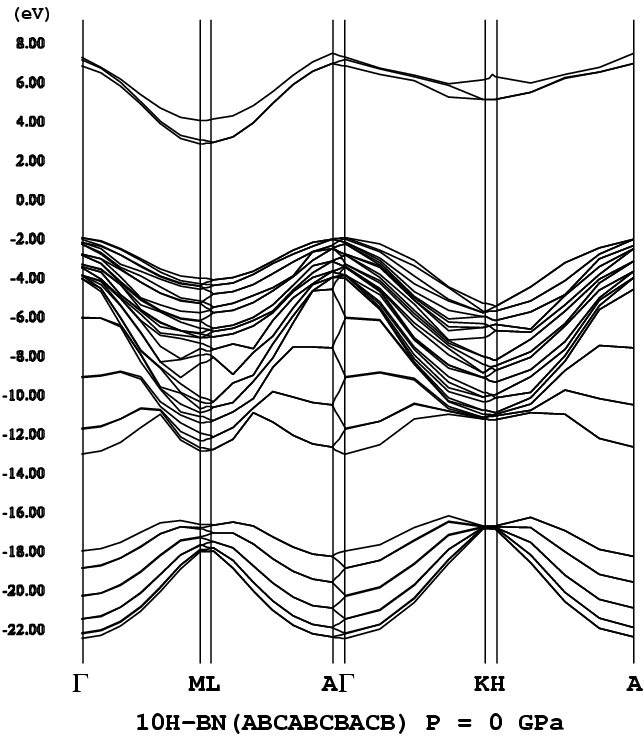
- [10H-BN](ABCABCBACB [H = 20%], P63mc, png[46 kb])
- [5H-BN](P3m1, png[40 kb])
- [4H-BN](P63mc, wide, png[63.5 kb])
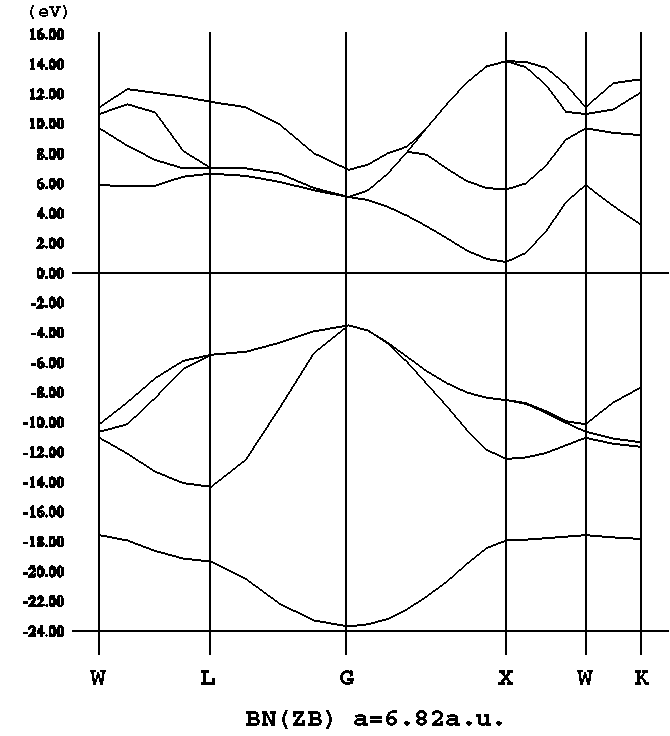
- [c-BN](3C-BN, png[7 kb])
- [3H-BN](= 3C-BN, P3m1, png[33.3 kb])
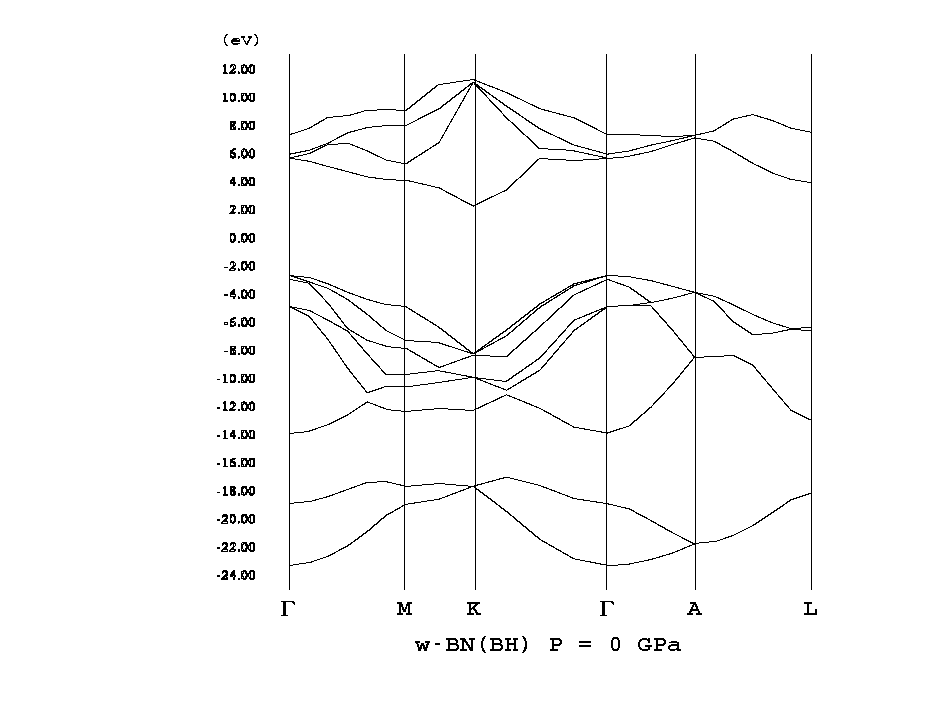
- [w-BN](2H-BN, png[7.6 kb])
- [2H-BN](w-BN, P63mc, wide, png[27.8 kb])
- [h-BN](A-B stacking, P63/mmc, png[26 kb])
- [h-BN](A-A stacking, P6_m2, png[23.4 kb])
- [h-BN](type B, P63/mmc, png[26 kb])
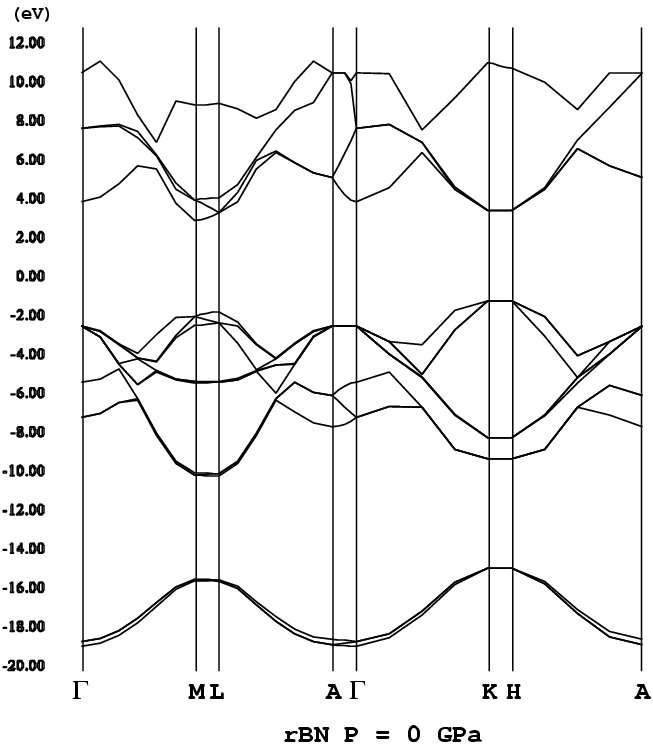
- [r-BN](rhombohedral BN, ABC stacking, png[28 kb])
- [Elctronic band structures of AlN polytypes]
- [10H-AlN](ABCBCBCBCB [H = 80%], P3m1, png[43.7 kb])
- [6H-AlN] as ABCACB [P63mc] and ABCBCB [P3m1] (png[173 kb])
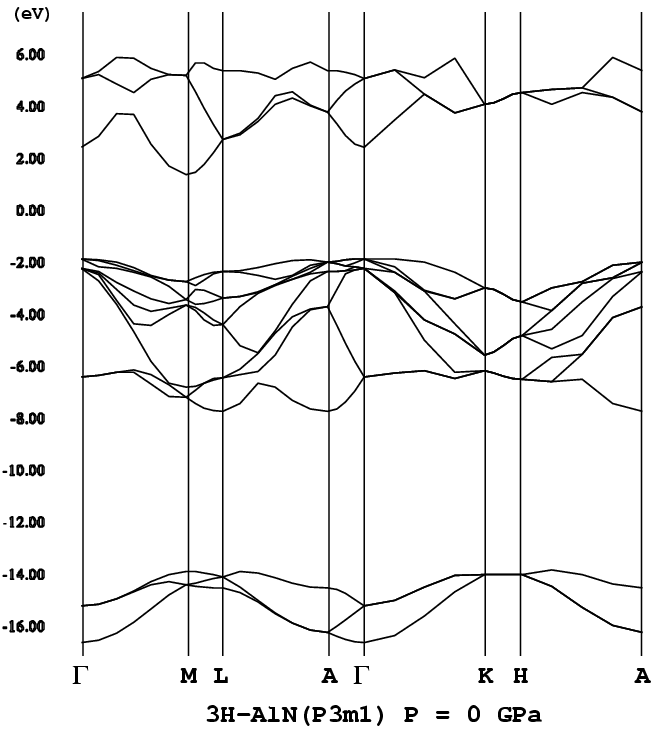
- [3H-AlN](= 3C-AlN, P3m1, png[27 kb])
- [2H-AlN](P63mc, png[24.3 kb])
- [Elctronic band structures of SiC polytypes]
- [4H-SiC](P63mc, wide, png[36 kb])
- [10H-SiC](ABCACBCACB [H = 40%], P3m1, png[56 kb])
-
- [関連国際会議]
- International Conference on Silicon Carbide and Related Materials 2026 (ICSCRM2026)、9月27日〜10月2日(2026)、開催地:パシフィコ横浜(横浜市、神奈川)、詳細は案内ページ参照。
-
- [関連ウェブページ]
- [BN compounds]
- [BC-compounds]
- [LiBC]
-
- [有用なウェブページ]
- Close-packed structures by P. Krishna and D. Pandey: International Union of CRYSTALLOGRAPHY(IUCr)[ポリタイプ構造に関しての有用な解説あり]
-
- POLYTYPISM IN SILICON CARBIDE by Dr. J. F. Kelly(Industrial Materials Group of Birkbeck College, University of London)[SiCポリタイプに関しての有用な解説]
-
- [BNに関する関連文献]
- [R1] O. Mishima and K. Era: in Electric Refractory Materials, ed. Y. Kumashiro (Marcel Dekker, New York, 2000) pp. 495-556, and references therein.
-
- [R2] S. Komatsu: J. Phys. D: Appl. Phys. 40 (2007) 2320 [Review].
- [R3] S. Komatsu, Y. Sato, D. Hirano, T. Nakamura, K. Koga, A. Yamamoto, T. Nagata, T. Chikyo, T. Watanabe, and T. Takizawa: Journal of Physics D: Applied Physics 42 (2009) 225107[P-type sp3-bonded BN/n-type Si][Heterodiode][Solar cell][Laser-plasma synchronous CVD method].
-
- [Ra] F. Oba, A. Togo, I. Tanaka, K. Watanabe, and T. Taniguchi, Phys. Rev. B81, 075125(2010)[VASP-PAW][Doping of h-BN][Intercalation][Prediction].
-
- [LFS] L. Liu, Y. P. Feng, and Z. X. Shen: Phys. Rev. B68 (2003) 104102 [h-BN].
-
- [ADDF]H. Tokoyama, H. Yamakado and K. Ohno, Chemistry Letters (doi:10.1246/cl.151114)(2015)[Automated exploration][ADDF][GRRM]
- [最近のh-BN関連論文]
- [h-BN][Layered BN] B. Huang, X. K. Cao, H. X. Jiang, J. Y. Lin and S.-H. Wei, Phys. Rev. B86, 155202(2012)[Significantly enhanced optical transition].
- [h-BN] S.-P. Gao, Solid State Communications 152 (2012) 1817[Band gap characters][Dispersion corrected][GW].
- [h-BN] G. Cassabois, P. Valvin, B. Gil, "Hexagonal boron nitride is an indirect bandgap semiconductor", arXiv:1512.02962.
- [h-BN] S. M. Gilbert, T. Pham, M. Dogan, S. Oh, B. Shevitski, G. Schumm, S. Liu, P. Ercius, S. Aloni, M. L. Cohen, A. Zettl, "Alternative Stacking Sequences in Hexagonal Boron Nitride", arXiv:1810.04814.
-
- [Polytype (Polytypism)に関する参考文献]
- [ITC]International Table for Crystallography, ed. A. J. C. Wilson and E. Prince (Kluwer, Dordrecht, 2004) Vol. C, Chap. 9.2 by S. Durovic, P. Krishna ans D. Pandey, pp. 744 - 765.
- [VK]A. R. Verma and P. Krishna: Polymorphism and Polytypism in Crystals (Wiley, New York, 1966).
- [Ig] J. E. Iglesias: Acta. Cryst. A62(2006) 178.
- A. L. Patterson and J. S. Kasper: ``International Tables for X-ray Crystallography'', ed. J. S. Kasper and K. Lonsdale (The Kynoch Press, Birmingham, 1967) Vol. 2, pp. 342 - 354.
- T. J. McLarnan: Z. Kristallogr. 155 (1981) 269.
- Z. Inoue: J. Mater. Sci. 17 (1982) 3189.
- A. L. Ortiz, F. Sanchez-Bajo, F. L. Cumbrera and F. Guiberteau, J. Appl. Cryst. 46 (2013) 242 -247[Prolific polytypism][SiC].
-
- [BN, SiC, AlN及び関連化合物に関する参考文献]
- Z. Pan, H. Sun, Yi Zhang and C. Chen: Phys. Rev. Lett., Vol. 102, No. 5, 055503(2009)[Harder than diamond][Hexagonal diamond, w-BN][PARATEC].
-
- L. Zhou, X. Ni, Ü. Özgür, H. Morkoç, R. P. Devaty, W. J. Choyke and D. J. Smith: Journal of Crystal Growth, Vol. 311, Issue 6 (2009) 1456-1459 [6H-AlN, m-plane AlN/SiC interface, Experiment]. <-- Consistent with our resutls for 6H-AlN polytypes [3].
-
- [C][Si][SiC][BN][AlN][GaN][InN]K. Moriguchi, K. Kamei, K. Kusunoki, N. Yashiro and N. Okada, J. Mater. Res., Vol. 28 (2013) 7-16[Comparative studies][Nonequivalent hexagonal polytypes].
- [関連論文](IUCr)
- [関連コード](テキスト形式、上記関連論文著者によるコード、有用、IUCr electronic archives[Reference: ZM5042])
-
- [Floating Electron States]Y. Matsushita, S. Furuya and A. Oshiyama, Phys. Rev. Lett., Vol. 108, No. 24, 246404(2012)[Covalent semiconductor][SiC][GaN][AlN][BN][Si][C].
- [Interstitial Channels]Y. Matsushita and A. Oshiyama, Phys. Rev. Lett., Vol. 112, No. 13, 136403(2014)[Control band gaps][Effective mass][Tetrahedrally bonded][SiC].
- [Electron Confinement Due to Stacking Control of Atomic Layers]Y. Matsushita, S. Furuya and A. Oshiyama, J. Phys. Soc. Jpn. 83 (2014) 094713[RSDFT][SiC][Roles of floating states][Spontaneous polarization].
- [Electronic states floating]Y. Matsushita A. Oshiyama, J. Phys. Soc. Jpn. 86 (2017) 054702[Comprehensive study][Band-gap variation][sp3-bonded semiconductor][Electronic states floating][Internal space].
- [Design and formation of SiC(0001)/SiO2 interfaces]T. Kobayashi, T. Okuda, K. Tachiki, K. Ito, Y. Matsushita and T. Kimoto, Appl. Phys. Express 13 091003 (2020)[Si deposition][Low-temperature oxidation][High-temperature nitridation].
- [Electron and Hole Confinement in Hetero-Crystalline SiC Superlattice]Y. Sugihara, K. Uchida and A. Oshiyama, J. Phys. Soc. Jpn. 84 (2015) 084709.
-
- [SiC][A simple approach to the polytypism in SiC]T. Ito, T. Akiyama and K. Nakamura, Journal of Crystal Growth 362, 207-210(2013).
- [SiC][Systematic theoretical investigations][Vacancy][N substitution]T. Ito, T. Akiyama and K. Nakamura, Phys. Status Solidi C (2013) in press.
-
- [10H-SiC]S. Nakashima, T. Tomita, N. Kuwahara, T. Mitani, M. Tomobe, S. Nishizawa and H. Okumura, J. Appl. Phys. 114, 193510(2013)[Raman intensity profiles][Zone-folded modes][3322]
-
- [SiC]S. Kawanishi, T. Mizoguchi, arXiv:1512.04626[Effect of van der Waals interactions][Stability]
-
- [SiC]M. Uemoto, N. Komatsu, Y. Egami and T. Ono, J. Phys. Soc. Jpn. 90, 124713 (2021)[First-principles study][Structure][Anisotropy][High N-atom density layer][4H-SiC]
-
- [STAM][Superconductor][SiC]Science and Technology of Advanced Materials(STAM)特集号 ("Focus on Superconductivity in Semiconductors")、Vol.9, Issue 4(2008). ←[SiC関連(超伝導)の記事あり]
-
- [C1][AlN]Y. C. Cheng, H. T. Chen, X. X. Li, X. L. Wu, J. Zhu, S. H. Li, and Paul K. Chu: Journal of Applied Physics 105 (2009) 083511[CASTEP][PWSCF][2H-,4H- and 6H-AlN][Optical and vibrational properties].
- [C2][AlN]Y. C. Cheng, X. L. Wu, S. H. Li, and Paul K. Chu: Applied Physics Letters 95 (2009) 121902[QUANTUM-ESPRESSO][4H-AlN][Stress influence][Band-edge luminescence].
-
- [その他の文献]
- [CP] R. Car and M. Parrinello, Phys. Rev. Lett., 55, 2471(1985).
- [BH] U. von Barth and L. Hedin, J. Phys. C5, 1629(1972).
- [Wigner] E. Wigner, Phys. Rev. 46, 1002(1934).
- [PBE] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865(1996).
- [TM] N. Troullier and J. L. Martins, Solid State Commun., 74, 613(1990); and Phys. Rev. B43, 1993(1991).
- [BHS] G. B. Bachelet, D. R. Hamann and M. Schlüter, Phys. Rev. B26, 4199(1982).
- [PCC] S. G. Louie, S. Froyen and M. L. Cohen, Phys. Rev. B26, 1738(1982).
- [KB] L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 48, 1425(1982).
- [LN] Z. Liu and J. Ni: J. Phys.: Condens. Matter 17 (2005) 5355.
- [PCLC] C. H. Park, B. Cheong, K. Lee, and K. J. Chang: Phys. Rev. B 49 (1994) 4485.
- [OBU]宇田毅、基礎講座「<半導体材料・プロセスの物理と設計>半導体バルクの構造と電子状態」、応用物理、第68巻、第7号、817頁(1999)←閃亜鉛鉱構造、ウルツ鉱構造における、イオン性、共有結合性と第三隣接原子同士との関係の記述(9節)あり。
[文頭][English][Top][E-mail]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}